ENCYCLOPEDIC ENTRY
Genetic variation.
Genetic variation is the presence of differences in sequences of genes between individual organisms of a species. It enables natural selection, one of the primary forces driving the evolution of life.
Biology, Genetics

genetic variation
In many species, special genetic variations give animals a camouflaged appearance to blend in with their environment, like this Catalpa Sphinx moth (Ceratomia catalpae) which uses its textured wings to blend in with a tree's bark.
Photograph by J.S. Houser

Genetic variation refers to differences among the genomes of members of the same species. A genome is all the hereditary information—all the genes —of an organism . For instance, the human genome contains somewhere between twenty and twenty-five thousand genes .
Genes are units of hereditary information, and they carry instructions for building proteins. The genes that are encoded within these proteins are what enable cells to function. Most organisms that reproduce sexually have two copies of each gene , because each parent cell or organism donates a single copy of its genes to its offspring. Additionally, genes can exist in slightly different forms, called alleles , which further adds to genetic variation.
The combination of alleles of a gene that an individual receives from both parents determines what biologists call the genotype for a particular trait, such as hair texture. The genotype that an individual possesses for a trait, in turn, determines the phenotype —the observable characteristics—such as whether that individual actually ends up with straight, wavy, or curly hair.
Genetic variation within a species can result from a few different sources. Mutations , the changes in the sequences of genes in DNA , are one source of genetic variation. Another source is gene flow , or the movement of genes between different groups of organisms . Finally, genetic variation can be a result of sexual reproduction , which leads to the creation of new combinations of genes .
Genetic variation in a group of organisms enables some organisms to survive better than others in the environment in which they live. Organisms of even a small population can differ strikingly in terms of how well suited they are for life in a certain environment . An example would be moths of the same species with different color wings. Moths with wings similar to the color of tree bark are better able to camouflage themselves than moths of a different color. As a result, the tree-colored moths are more likely to survive, reproduce, and pass on their genes . This process is called natural selection , and it is the main force that drives evolution .
Media Credits
The audio, illustrations, photos, and videos are credited beneath the media asset, except for promotional images, which generally link to another page that contains the media credit. The Rights Holder for media is the person or group credited.
Production Managers
Program specialists, last updated.
October 19, 2023
User Permissions
For information on user permissions, please read our Terms of Service. If you have questions about how to cite anything on our website in your project or classroom presentation, please contact your teacher. They will best know the preferred format. When you reach out to them, you will need the page title, URL, and the date you accessed the resource.
If a media asset is downloadable, a download button appears in the corner of the media viewer. If no button appears, you cannot download or save the media.
Text on this page is printable and can be used according to our Terms of Service .
Interactives
Any interactives on this page can only be played while you are visiting our website. You cannot download interactives.
Related Resources
Genetic Variation Definition, Causes, and Examples
Japatino / Moment Open / Getty Images
- Cell Biology
- Weather & Climate
- B.A., Biology, Emory University
- A.S., Nursing, Chattahoochee Technical College
Genetic variation can be defined as the genetic makeup of organisms within a population change. Genes are inherited segments of DNA that contain codes for the production of proteins. Genes exist in alternate versions, or alleles , that determine distinct traits that can be passed on from parents to offspring.
Key Takeaways: Genetic Variation
- Genetic variation refers to differences in the genetic makeup of individuals in a population.
- Genetic variation is necessary in natural selection . In natural selection, organisms with environmentally selected traits are better able to adapt to the environment and pass on their genes.
- Major causes of variation include mutations, gene flow, and sexual reproduction.
- DNA mutation causes genetic variation by altering the genes of individuals in a population.
- Gene flow leads to genetic variation as new individuals with different gene combinations migrate into a population.
- Sexual reproduction promotes variable gene combinations in a population leading to genetic variation.
- Examples of genetic variation include eye color, blood type, camouflage in animals, and leaf modification in plants.
Genetic variation is important to the processes of natural selection and biological evolution . The genetic variations that arise in a population happen by chance, but the process of natural selection does not. Natural selection is the result of the interactions between genetic variations in a population and the environment. The environment determines which genetic variations are more favorable or better suited for survival. As organisms with these environmentally selected genes survive and reproduce, more favorable traits are passed on to the population as a whole.
Genetic Variation Causes
Alfred Pasieka / Science Photo Library / Getty Images
Genetic variation occurs mainly through DNA mutation , gene flow (movement of genes from one population to another), and sexual reproduction . Due to the fact that environments are unstable, populations that are genetically variable will be able to adapt to changing situations better than those that do not contain genetic variation.
- DNA Mutation : A mutation is a change in the DNA sequence. These variations in gene sequences can sometimes be advantageous to an organism. Most mutations that result in genetic variation produce traits that confer neither an advantage or disadvantage. Mutations lead to genetic variation by altering genes and alleles in a population. They may impact an individual gene or an entire chromosome. Although mutations change an organism's genotype (genetic makeup), they may not necessarily change an organism's phenotype .
- Gene Flow: Also called gene migration, gene flow introduces new genes into a population as organisms migrate into a new environment. New gene combinations are made possible by the availability of new alleles in the gene pool. Gene frequencies may also be altered by the emigration of organisms out of a population. The immigration of new organisms into a population may help organisms better adapt to changing environmental conditions. The migration of organisms out of a population could result in a lack of genetic diversity.
- Sexual Reproduction: Sexual reproduction promotes genetic variation by producing different gene combinations. Meiosis is the process by which sex cells or gametes are created. Genetic variation occurs as alleles in gametes are separated and randomly united upon fertilization . The genetic recombination of genes also occurs during crossing over or the swapping of gene segments in homologous chromosomes during meiosis.
Genetic Variation Examples
David G Richardson / Getty Images
Favorable genetic traits in a population are determined by the environment. Organisms that are better able to adapt to their environment survive to pass on their genes and favorable traits. Sexual selection is commonly seen in nature as animals tend to select mates that have traits that are favorable. As females mate more often with males considered to have more favorable traits, these genes occur more often in a population over time.
A person's skin color , hair color, dimples, freckles, and blood type are all examples of genetic variations that can occur in a human population . Examples of genetic variation in plants include the modified leaves of carnivorous plants and the development of flowers that resemble insects to lure plant pollinators . Gene variation in plants often occurs as the result of gene flow. Pollen is dispersed from one area to another by the wind or by pollinators over great distances.
Examples of genetic variation in animals include albinism, cheetahs with stripes, snakes that fly , animals that play dead , and animals that mimic leaves . These variations enable the animals to better adapt to conditions in their environments.
- 5 Conditions for Hardy-Weinberg Equilibrium
- 6 Things You Should Know About Biological Evolution
- Genes and Genetic Inheritance
- Phenotype: How a Gene Is Expressed As a Physical Trait
- Genetics Basics
- Causes of Microevolution
- An Introduction to Evolution
- Asexual vs. Sexual Reproduction
- Descent with Modification
- What Is Pleiotropy? Definition and Examples
- What Is Selective Sweep?
- 4 Necessary Factors for Natural Selection
- Chromosome Mutations
- 5 Types of Asexual Reproduction
- 4 Types of Reproduction
- Genetic Drift

- school Campus Bookshelves
- menu_book Bookshelves
- perm_media Learning Objects
- login Login
- how_to_reg Request Instructor Account
- hub Instructor Commons
Margin Size
- Download Page (PDF)
- Download Full Book (PDF)
- Periodic Table
- Physics Constants
- Scientific Calculator
- Reference & Cite
- Tools expand_more
- Readability
selected template will load here
This action is not available.
19.2A: Genetic Variation
- Last updated
- Save as PDF
- Page ID 13482

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
Genetic variation is a measure of the variation that exists in the genetic makeup of individuals within population.
Learning Objectives
- Assess the ways in which genetic variance affects the evolution of populations
- Genetic variation is an important force in evolution as it allows natural selection to increase or decrease frequency of alleles already in the population.
- Genetic variation can be caused by mutation (which can create entirely new alleles in a population), random mating, random fertilization, and recombination between homologous chromosomes during meiosis (which reshuffles alleles within an organism’s offspring).
- Genetic variation is advantageous to a population because it enables some individuals to adapt to the environment while maintaining the survival of the population.
- genetic diversity : the level of biodiversity, refers to the total number of genetic characteristics in the genetic makeup of a species
- crossing over : the exchange of genetic material between homologous chromosomes that results in recombinant chromosomes
- phenotypic variation : variation (due to underlying heritable genetic variation); a fundamental prerequisite for evolution by natural selection
- genetic variation : variation in alleles of genes that occurs both within and among populations
Genetic Variation
Genetic variation is a measure of the genetic differences that exist within a population. The genetic variation of an entire species is often called genetic diversity. Genetic variations are the differences in DNA segments or genes between individuals and each variation of a gene is called an allele.For example, a population with many different alleles at a single chromosome locus has a high amount of genetic variation. Genetic variation is essential for natural selection because natural selection can only increase or decrease frequency of alleles that already exist in the population.
Genetic variation is caused by:
- random mating between organisms
- random fertilization
- crossing over (or recombination) between chromatids of homologous chromosomes during meiosis
The last three of these factors reshuffle alleles within a population, giving offspring combinations which differ from their parents and from others.

Evolution and Adaptation to the Environment
Variation allows some individuals within a population to adapt to the changing environment. Because natural selection acts directly only on phenotypes, more genetic variation within a population usually enables more phenotypic variation. Some new alleles increase an organism’s ability to survive and reproduce, which then ensures the survival of the allele in the population. Other new alleles may be immediately detrimental (such as a malformed oxygen-carrying protein) and organisms carrying these new mutations will die out. Neutral alleles are neither selected for nor against and usually remain in the population. Genetic variation is advantageous because it enables some individuals and, therefore, a population, to survive despite a changing environment.

Geographic Variation
Some species display geographic variation as well as variation within a population. Geographic variation, or the distinctions in the genetic makeup of different populations, often occurs when populations are geographically separated by environmental barriers or when they are under selection pressures from a different environment. One example of geographic variation are clines: graded changes in a character down a geographic axis.
Sources of Genetic Variation
Gene duplication, mutation, or other processes can produce new genes and alleles and increase genetic variation. New genetic variation can be created within generations in a population, so a population with rapid reproduction rates will probably have high genetic variation. However, existing genes can be arranged in new ways from chromosomal crossing over and recombination in sexual reproduction. Overall, the main sources of genetic variation are the formation of new alleles, the altering of gene number or position, rapid reproduction, and sexual reproduction.
Thank you for visiting nature.com. You are using a browser version with limited support for CSS. To obtain the best experience, we recommend you use a more up to date browser (or turn off compatibility mode in Internet Explorer). In the meantime, to ensure continued support, we are displaying the site without styles and JavaScript.
- View all journals
- Explore content
- About the journal
- Publish with us
- Sign up for alerts
- Review Article
- Published: April 2009
Human genetic variation and its contribution to complex traits
- Kelly A. Frazer 1 ,
- Sarah S. Murray 1 ,
- Nicholas J. Schork 1 &
- Eric J. Topol 1
Nature Reviews Genetics volume 10 , pages 241–251 ( 2009 ) Cite this article
18k Accesses
726 Citations
23 Altmetric
Metrics details
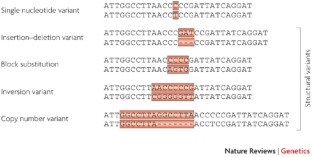
Human genetic variants are typically referred to as either common or rare, to denote the frequency of the minor allele in the human population. Genetic variants can also be divided into two different nucleotide composition classes — single nucleotide variants and structural variants.
The alleles of SNPs located in the same genomic interval are often correlated with one another. This correlation structure, or linkage disequilibrium (LD), varies in a complex and unpredictable manner across the genome and between different populations.
Structural variants seem to behave similarly to SNPs in terms of both genomic and population distribution, indicating a similar evolutionary history: both types of variants are 'ancestral' having arisen once in human history and then shared among individuals by descent rather than being the result of recurrent mutations.
Full sequencing of human genomes has shown that in any given individual there are, on average, ∼ 4 million genetic variants encompassing ∼ 12 Mb of sequence. The challenge is to determine which of these variants underlie or are responsible for the inherited components of phenotypes.
Over the last decade or so the human genetics field has debated the common disease–common variant hypothesis, which posits that common complex traits are largely due to common variants with small-to-modest affect sizes. The opposing theory, the rare variant hypothesis, posits that common complex traits are the summation of low-frequency, high-penetrance variants.
Genome-wide association (GWA) studies are the most widely used contemporary approach to relate genetic variation to phenotypic diversity. Over the past 2 years these studies have identified statistical association between hundreds of loci across the genome and common complex traits.
Most of the genes or genomic loci that have been identified by GWA studies have not previously been known to be related to the complex trait under investigation. Surprisingly, there have been several instances in which one genomic interval has been associated with two or more seemingly distinct diseases.
An unforeseen limitation of GWA studies is that the genomic markers that are found to be associated with any given complex trait each have less impact on susceptibility than was anticipated. Most of the odds ratios for the heterozygote genotypes of the associated variants that have been identified so far are approximately 1.1, a figure that can increase to 1.5–1.6 for homozygote genotypes.
At this point, there are almost no complex traits for which more than 10% of the genetic variance is explained, and many are far below that threshold, leaving the bulk of heritability unexplained by the common variants identified so far.
One possibility is that the missing variation is accounted for by common genetic variants with small effect sizes that have not yet been identified. Some of the missing heritability is probably accounted for by rare and novel variants. Additionally, there are statistical limitations of the GWA approach in identifying gene–gene and gene–environment interactions, which are likely to be profoundly important.
The last few years have seen extensive efforts to catalogue human genetic variation and correlate it with phenotypic differences. Most common SNPs have now been assessed in genome-wide studies for statistical associations with many complex traits, including many important common diseases. Although these studies have provided new biological insights, only a limited amount of the heritable component of any complex trait has been identified and it remains a challenge to elucidate the functional link between associated variants and phenotypic traits. Technological advances, such as the ability to detect rare and structural variants, and a clear understanding of the challenges in linking different types of variation with phenotype, will be essential for future progress.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
176,64 € per year
only 14,72 € per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout
Similar content being viewed by others

A global overview of pleiotropy and genetic architecture in complex traits

The collective effects of genetic variants and complex traits

Genome-wide association studies
Lander, E. S. et al. Initial sequencing and analysis of the human genome. Nature 409 , 860–921 (2001).
Article CAS PubMed Google Scholar
Venter, J. C. et al. The sequence of the human genome. Science 291 , 1304–1351 (2001).
Altshuler, D. et al. A haplotype map of the human genome. Nature 437 , 1299–1320 (2005).
Google Scholar
Frazer, K. A. et al. A second generation human haplotype map of over 3.1 million SNPs. Nature 449 , 851–861 (2007). Publication of the HapMap Phase II results genotyping over 3.1 million SNPs in 270 individuals from four geographically diverse populations.
CAS PubMed Google Scholar
Hinds, D. A. et al. Whole-genome patterns of common DNA variation in three human populations. Science 307 , 1072–1079 (2005).
Sebat, J. et al. Large-scale copy number polymorphism in the human genome. Science 305 , 525–528 (2004).
Iafrate, A. J. et al. Detection of large-scale variation in the human genome. Nature Genet. 36 , 949–951 (2004).
Redon, R. et al. Global variation in copy number in the human genome. Nature 444 , 444–454 (2006).
CAS PubMed PubMed Central Google Scholar
Kidd, J. M. et al. Mapping and sequencing of structural variation from eight human genomes. Nature 453 , 56–64 (2008). Demonstrates the prevalence and importance of structural variation in the human genome, which historically had not been given much attention.
Eichler, E. E. et al. Completing the map of human genetic variation. Nature 447 , 161–165 (2007).
Levy, S. et al. The diploid genome sequence of an individual human. PLoS Biol. 5 , e254 (2007). The first publication of a genome sequence of a single individual (J. Craig Venter).
PubMed PubMed Central Google Scholar
Wheeler, D. A. et al. The complete genome of an individual by massively parallel DNA sequencing. Nature 452 , 872–876 (2008). The first paper to demonstrate how technological advances will enable the rapid sequencing of individual human genomes in the near future. Interestingly, the individual sequenced here is Jim Watson, who won the nobel prize for discovery of the DNA double helix.
Wang, J. et al. The diploid genome sequence of an Asian individual. Nature 456 , 60–65 (2008).
Bentley, D. R. et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456 , 53–59 (2008).
McCarthy, M. I. et al. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nature Rev. Genet. 9 , 356–369 (2008). A useful review of appropriate study design, analysis, and interpretation of human GWA studies.
Feuk, L., Carson, A. R. & Scherer, S. W. Structural variation in the human genome. Nature Rev. Genet. 7 , 85–97 (2006).
Conrad, D. F. & Hurles, M. E. The population genetics of structural variation. Nature Genet. 39 , S30–S36 (2007).
Altshuler, D., Daly, M. J. & Lander, E. S. Genetic mapping in human disease. Science 322 , 881–888 (2008). A good recent review of the results of human GWA studies. Interestingly, the authors compare sample size requirements for genetic association studies of common and rare variants.
Donnelly, P. Progress and challenges in genome-wide association studies in humans. Nature 456 , 728–731 (2008).
Kruglyak, L. The road to genome-wide association studies. Nature Rev. Genet. 9 , 314–318 (2008).
Kimura, M. Evolutionary rate at the molecular level. Nature 217 , 624–626 (1968).
Ohta, T. Near-neutrality in evolution of genes and gene regulation. Proc. Natl Acad. Sci. USA 99 , 16134–16137 (2002).
Kruglyak, L. & Nickerson, D. A. Variation is the spice of life. Nature Genet. 27 , 234–236 (2001).
Slatkin, M. Linkage disequilibrium — understanding the evolutionary past and mapping the medical future. Nature Rev. Genet. 9 , 477–485 (2008).
Barrett, J. C. & Cardon, L. R. Evaluating coverage of genome-wide association studies. Nature Genet. 38 , 659–662 (2006).
Eberle, M. A. et al. Power to detect risk alleles using genome-wide tag SNP panels. PLoS Genet. 3 , e170 (2007).
PubMed Central Google Scholar
Pe'er, I. et al. Evaluating and improving power in whole-genome association studies using fixed marker sets. Nature Genet. 38 , 663–667 (2006).
Clark, A. G. & Li, J. Conjuring SNPs to detect associations. Nature Genet. 39 , 815–816 (2007).
Tuzun, E. et al. Fine-scale structural variation of the human genome. Nature Genet. 37 , 727–732 (2005).
Cooper, G. M., Zerr, T., Kidd, J. M., Eichler, E. E. & Nickerson, D. A. Systematic assessment of copy number variant detection via genome-wide SNP genotyping. Nature Genet. 40 , 1199–1203 (2008).
Korbel, J. O. & al, e. Paired-end mapping reveals extensive structural variation in the human genome. Science 318 , 420–426 (2007).
Khaja, R. et al. Genome assembly comparison identifies structural variants in the human genome. Nature Genet. 38 , 1413–1418 (2006).
Conrad, D. F., Andrews, T. D., Carter, N. P., Hurles, M. E. & Pritchard, J. K. A high-resolution survey of deletion polymorphism in the human genome. Nature Genet. 38 , 75–81 (2006).
Barnes, C. et al. A robust statistical method for case-control association testing with copy number variation. Nature Genet. 40 , 1245–1252 (2008).
McCarroll, S. A. & Altshuler, D. M. Copy-number variation and association studies of human disease. Nature Genet. 39 , S37–S42 (2007).
Sebat, J. Major changes in our DNA lead to major changes in our thinking. Nature Genet. 39 , S3–S5 (2007).
Korn, J. M. et al. Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs. Nature Genet. 40 , 1253–1260 (2008).
Cooper, G. M., Nickerson, D. A. & Eichler, E. E. Mutational and selective effects on copy-number variants in the human genome. Nature Genet. 39 , S22–S29 (2007).
Hinds, D. A., Kloek, A. P., Jen, M., Chen, X. & Frazer, K. A. Common deletions and SNPs are in linkage disequilibrium in the human genome. Nature Genet. 38 , 9–11 (2006).
McCarroll, S. A. et al. Common deletion polymorphisms in the human genome. Nature Genet. 38 , 86–92 (2006).
McCarroll, S. A. et al. Integrated detection and population-genetic analysis of SNPs and copy number variation. Nature Genet. 40 , 1166–1174 (2008). Demonstrates that common structural variants are in LD with common SNPs in the human genome.
Jakobsson, M. et al. Genotype, haplotype and copy-number variation in worldwide human populations. Nature 451 , 998–1003 (2008).
Bailey, J. A. et al. Recent segmental duplications in the human genome. Science 297 , 1003–1007 (2002).
Locke, D. P. et al. Linkage disequilibrium and heritability of copy-number polymorphisms within duplicated regions of the human genome. Am. J. Hum. Genet. 79 , 275–290 (2006).
Pritchard, J. K. & Cox, N. J. The allelic architecture of human disease genes: common disease–common variant or not? Hum. Mol. Genet. 11 , 2417–2423 (2002).
Pritchard, J. K. Are rare variants responsible for susceptibility to complex diseases? Am. J. Hum. Genet. 69 , 124–137 (2001).
Reich, D. E. & Lander, E. S. On the allelic spectrum of human disease. Trends Genet. 17 , 502–510 (2001).
Lander, E. S. The new genomics: global views of biology. Science 274 , 536–539 (1996).
Chakravarti, A. Population genetics — making sense out of sequence. Nature Genet. 21 , 56–60 (1999).
Fearnhead, N. S., Winney, B. & Bodmer, W. F. Rare variant hypothesis for multifactorial inheritance: susceptibility to colorectal adenomas as a model. Cell Cycle 4 , 521–525 (2005).
Bodmer, W. & Bonilla, C. Common and rare variants in multifactorial susceptibility to common diseases. Nature Genet. 40 , 695–701 (2008). The authors discuss the concepts behind the common disease common–variant hypothesis and contrast them to the basic ideas that underlie the rare variant hypothesis.
Pearson, T. A. & Manolio, T. A. How to interpret a genome-wide association study. JAMA 299 , 1335–1344 (2008).
Iles, M. M. What can genome-wide association studies tell us about the genetics of common disease? PLoS Genet. 4 , e33 (2008).
Altshuler, D. & Daly, M. J. Guilt beyond a reasonable doubt. Nature Genet. 39 , 813–815 (2007).
Hindorff, L. A., Junkins, H. A., Mehta, J. P. and Manolio, T. A. A Catalog of Published Genome-Wide Association Studies. National Human Genome Research Institute [ online ], (accessed 1 Jan 2009).
Xavier, R. J. & Rioux, J. D. Genome-wide association studies: a new window into immune-mediated diseases. Nature Rev. Immunol. 8 , 631–643 (2008).
CAS Google Scholar
Frayling, T. M. Genome-wide association studies provide new insights into type 2 diabetes aetiology. Nature Rev. Genet. 8 , 657–662 (2007).
Lyssenko, V. et al. Common variant in MTNR1B associated with increased risk of type 2 diabetes and impaired early insulin secretion. Nature Genet. 41 , 82–88 (2009).
Bouatia-Naji, N. et al. A variant near MTNR1B is associated with increased fasting plasma glucose levels and type 2 diabetes risk. Nature Genet. 41 , 89–94 (2009).
Aulchenko, Y. S. et al. Genetic variation in the KIF1B locus influences susceptibility to multiple sclerosis. Nature Genet. 40 , 1402–1403 (2008).
Hafler, D. A. et al. Risk alleles for multiple sclerosis identified by a genomewide study. N. Engl. J. Med. 357 , 851–862 (2007).
Goh, K. I. et al. The human disease network. Proc. Natl Acad. Sci. USA 104 , 8685–8690 (2007).
Lettre, G. & Rioux, J. D. Autoimmune diseases: insights from genome-wide association studies. Hum. Mol. Genet. 17 , R116–R121 (2008).
McPherson, R. et al. A common allele on chromosome 9 associated with coronary heart disease. Science 316 , 1488–1491 (2007).
Samani, N. J. et al. Genomewide association analysis of coronary artery disease. N. Engl. J. Med. 357 , 443–453 (2007).
Helgadottir, A. et al. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science 316 , 1491–1493 (2007).
Helgadottir, A. et al. The same sequence variant on 9p21 associates with myocardial infarction, abdominal aortic aneurysm and intracranial aneurysm. Nature Genet. 40 , 217–224 (2008).
Amundadottir, L. T. et al. A common variant associated with prostate cancer in European and African populations. Nature Genet. 38 , 652–658 (2006).
Freedman, M. L. et al. Admixture mapping identifies 8q24 as a prostate cancer risk locus in African-American men. Proc. Natl Acad. Sci. USA 103 , 14068–14073 (2006).
Cookson, W. et al. Mapping complex disease traits with global gene expression. Nature Rev. Genet. 10 , 184–194 (2009).
Myles, S., Davison, D., Barrett, J., Stoneking, M. & Timpson, N. Worldwide population differentiation at disease-associated SNPs. BMC Med. Genomics 1 , 22 (2008).
Sladek, R. et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature 445 , 881–885 (2007).
Saxena, R. et al. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science 316 , 1331–1336 (2007).
Scott, L. J. et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science 316 , 1341–1345 (2007).
Zeggini, E. et al. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science 316 , 1336–1341 (2007).
Xing, J. et al. HapMap tagSNP transferability in multiple populations: general guidelines. Genomics 92 , 41–51 (2008).
Chanock, S. J. et al. Replicating genotype–phenotype associations. Nature 447 , 655–660 (2007).
Weiss, L. A. et al. Association between microdeletion and microduplication at 16p11.2 and autism. N. Engl. J. Med. 358 , 667–675 (2008).
Kumar, R. A. et al. Recurrent 16p11.2 microdeletions in autism. Hum. Mol. Genet. 17 , 628–638 (2008).
Marshall, C. R. et al. Structural variation of chromosomes in autism spectrum disorder. Am. J. Hum. Genet. 82 , 477–488 (2008).
Stefansson, H. et al. Large recurrent microdeletions associated with schizophrenia. Nature 455 , 232–236 (2008).
Consortium, I. S. Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature 455 , 237–241 (2008).
Walsh, T. et al. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science 320 , 539–543 (2008).
Richards, J. B. et al. Male-pattern baldness susceptibility locus at 20p11. Nature Genet. 40 , 1282–1284 (2008).
Link, E. et al. SLCO1B1 variants and statin-induced myopathy — a genomewide study. N. Engl. J. Med. 359 , 789–799 (2008).
Graham, R. R. et al. Genetic variants near TNFAIP3 on 6q23 are associated with systemic lupus erythematosus. Nature Genet. 40 , 1059–1061 (2008).
Barrett, J. C. et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nature Genet. 40 , 955–962 (2008). One of the human traits for which a large number of loci has been identified; the majority have modest effect sizes and in sum explain only a minority of the overall heritability.
Sulem, P. et al. Two newly identified genetic determinants of pigmentation in Europeans. Nature Genet. 40 , 835–837 (2008).
Sulem, P. et al. Genetic determinants of hair, eye and skin pigmentation in Europeans. Nature Genet. 39 , 1443–1452 (2007).
Stokowski, R. P. et al. A genomewide association study of skin pigmentation in a South Asian population. Am. J. Hum. Genet. 81 , 1119–1132 (2007).
Buch, S. et al. A genome-wide association scan identifies the hepatic cholesterol transporter ABCG8 as a susceptibility factor for human gallstone disease. Nature Genet. 39 , 995–999 (2007).
Klein, R. J. et al. Complement factor H polymorphism in age-related macular degeneration. Science 308 , 385–389 (2005).
Thorleifsson, G. et al. Common sequence variants in the LOXL1 gene confer susceptibility to exfoliation glaucoma. Science 317 , 1397–1400 (2007).
Weedon, M. N. et al. A common variant of HMGA2 is associated with adult and childhood height in the general population. Nature Genet. 39 , 1245–1250 (2007).
Sanna, S. et al. Common variants in the GDF5-UQCC region are associated with variation in human height. Nature Genet. 40 , 198–203 (2008).
Weedon, M. N. et al. Genome-wide association analysis identifies 20 loci that influence adult height. Nature Genet. 40 , 575–583 (2008).
Lettre, G. et al. Identification of ten loci associated with height highlights new biological pathways in human growth. Nature Genet. 40 , 584–591 (2008).
Gudbjartsson, D. F. et al. Many sequence variants affecting diversity of adult human height. Nature Genet. 40 , 609–615 (2008).
Weedon, M. N. & Frayling, T. M. Reaching new heights: insights into the genetics of human stature. Trends Genet. 24 , 595–603 (2008).
Aulchenko, Y. S. et al. Loci influencing lipid levels and coronary heart disease risk in 16 European population cohorts. Nature Genet. 41 , 47–55 (2008).
PubMed Google Scholar
Willer, C. J. et al. Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nature Genet. 40 , 161–169 (2008).
Kathiresan, S. et al. Common variants at 30 loci contribute to polygenic dyslipidemia. Nature Genet. 41 , 56–65 (2009).
Fearnhead, N. S. et al. Multiple rare variants in different genes account for multifactorial inherited susceptibility to colorectal adenomas. Proc. Natl Acad. Sci. USA 101 , 15992–15997 (2004).
Cohen, J. C. et al. Multiple rare alleles contribute to low plasma levels of HDL cholesterol. Science 305 , 869–872 (2004). One of the first studies to demonstrate that multiple rare alleles with high penetrance collectively contribute to a common phenotype in the general population.
Kotowski, I. K. et al. A spectrum of PCSK9 alleles contributes to plasma levels of low-density lipoprotein cholesterol. Am. J. Hum. Genet. 78 , 410–422 (2006).
Romeo, S. et al. Population-based resequencing of ANGPTL4 uncovers variations that reduce triglycerides and increase HDL. Nature Genet. 39 , 513–516 (2007).
Marini, N. J. et al. The prevalence of folate-remedial MTHFR enzyme variants in humans. Proc. Natl Acad. Sci. USA 105 , 8055–8060 (2008).
Birney, E. et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447 , 799–816 (2007). The goal of this project was to develop efficient methods for functionally annotating human genomic sequences. The work yielded new understandings of transcription regulatory sequences and their relationships with features of chromatin accessibility and histone modification.
Wade, C. M. & Daly, M. J. Genetic variation in laboratory mice. Nature Genet. 37 , 1175–1180 (2005).
Erickson, R. P. Mouse models of human genetic disease: which mouse is more like a man? Bioessays 18 , 993–998 (1996).
Linder, C. C. The influence of genetic background on spontaneous and genetically engineered mouse models of complex diseases. Lab. Anim. (NY) 30 , 34–39 (2001).
Frankel, W. N. Taking stock of complex trait genetics in mice. Trends Genet. 11 , 471–477 (1995).
Shao, H. et al. Genetic architecture of complex traits: large phenotypic effects and pervasive epistasis. Proc. Natl Acad. Sci. USA 105 , 19910–19914 (2008).
Maller, J. et al. Common variation in three genes, including a noncoding variant in CFH , strongly influences risk of age-related macular degeneration. Nature Genet. 38 , 1055–1059 (2006).
Li, M. et al. CFH haplotypes without the Y402H coding variant show strong association with susceptibility to age-related macular degeneration. Nature Genet. 38 , 1049–1054 (2006).
Rioux, J. D. et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nature Genet. 39 , 596–604 (2007).
Ng, P. C. et al. Genetic variation in an individual human exome. PLoS Genet. 4 , e1000160 (2008).
Carlson, C. S. et al. Selecting a maximally informative set of single-nucleotide polymorphisms for association analyses using linkage disequilibrium. Am. J. Hum. Genet. 74 , 106–120 (2004).
Pollin, T. I. et al. A null mutation in human APOC3 confers a favorable plasma lipid profile and apparent cardioprotection. Science 322 , 1702–1705 (2008).
Sabatti, C. et al. Genome-wide association analysis of metabolic traits in a birth cohort from a founder population. Nature Genet. 41 , 35–46 (2008).
Scherer, S. W. et al. Challenges and standards in integrating surveys of structural variation. Nature Genet. 39 , S7–S15 (2007).
Stefansson, H. et al. A common inversion under selection in Europeans. Nature Genet. 37 , 129–137 (2005).
Zeggini, E. et al. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nature Genet. 40 , 638–645 (2008).
Steinthorsdottir, V. et al. A variant in CDKAL1 influences insulin response and risk of type 2 diabetes. Nature Genet. 39 , 770–775 (2007).
Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447 , 661–678 (2007).
Frayling, T. M. et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science 316 , 889–894 (2007).
Gerken, T. et al. The obesity-associated FTO gene encodes a 2-oxoglutarate-dependent nucleic acid demethylase. Science 318 , 1469–1472 (2007).
Prokopenko, I. et al. Variants in MTNR1B influence fasting glucose levels. Nature Genet. 41 , 77–81 (2009).
Bayat, A., Barton, A. & Ollier, W. E. Dissection of complex genetic disease: implications for orthopaedics. Clin. Orthop. Relat. Res. 419 , 297–305 (2004).
Vang, T. et al. Autoimmune-associated lymphoid tyrosine phosphatase is a gain-of-function variant. Nature Genet. 37 , 1317–1319 (2005).
Gudmundsson, J. et al. Two variants on chromosome 17 confer prostate cancer risk, and the one in TCF2 protects against type 2 diabetes. Nature Genet. 39 , 977–983 (2007).
Thomas, G. et al. Multiple loci identified in a genome-wide association study of prostate cancer. Nature Genet. 40 , 310–315 (2008).
Download references
Acknowledgements
The authors are supported by a National Institutes of Health grant (NIH 1U54RR025204-01).
Author information
Authors and affiliations.
Scripps Genomic Medicine, Scripps Translational Science Institute and The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, 92037, California, USA
Kelly A. Frazer, Sarah S. Murray, Nicholas J. Schork & Eric J. Topol
You can also search for this author in PubMed Google Scholar
Corresponding author
Correspondence to Kelly A. Frazer .
Related links
Further information.
The Scripps Research Institute
International HapMap Project
Perlegen Sciences
Broadly defined, these are all variants that are not single nucleotide variants. They include insertion–deletions, block substitutions, inversions of DNA sequences and copy number differences.
An investigation of the association between common genetic variation and disease. This type of analysis requires a dense set of markers (for example, SNPs) that capture a substantial proportion of common variation across the genome, and large numbers of study subjects.
Continuously distributed phenotypes that are classically believed to result from the independent action of many genes, environmental factors and gene-by-environment interactions.
The less common allele of a polymorphism.
(LD). In population genetics, LD is the nonrandom association of alleles. For example, alleles of SNPs that reside near one another on a chromosome often occur in nonrandom combinations owing to infrequent recombination.
Subdivision of a population into different ethnic groups with potentially different marker allele frequencies and different disease prevalences.
A measurement of association that is commonly used in case–control studies. It is defined as the odds of exposure to the susceptible genetic variant in cases compared with that in controls. If the odds ratio is significantly greater than one, then the genetic variant is associated with the disease.
In statistical genetics, this term refers to an interaction of multiple genetic variants (usually at different loci) such that the net phenotypic effect of carrying more than one variant is different than would be predicted by simply combining the effects of each individual variant.
Rights and permissions
Reprints and permissions
About this article
Cite this article.
Frazer, K., Murray, S., Schork, N. et al. Human genetic variation and its contribution to complex traits. Nat Rev Genet 10 , 241–251 (2009). https://doi.org/10.1038/nrg2554
Download citation
Issue Date : April 2009
DOI : https://doi.org/10.1038/nrg2554
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
This article is cited by
E-gwas: an ensemble-like gwas strategy that provides effective control over false positive rates without decreasing true positives.
- Guang-Liang Zhou
- Fang-Jun Xu
- Meng-Jin Zhu
Genetics Selection Evolution (2023)
Gene-environment interaction explains a part of missing heritability in human body mass index
- Hae-Un Jung
- Dong Jun Kim
- Bermseok Oh
Communications Biology (2023)
Weighted kernels improve multi-environment genomic prediction
- Brett F. Carver
- Charles Chen
Heredity (2023)
Machine learning techniques for pathogenicity prediction of non-synonymous single nucleotide polymorphisms in human body
- Enas M. F. El Houby
Journal of Ambient Intelligence and Humanized Computing (2023)
Artificial Intelligence Techniques for the effective diagnosis of Alzheimer’s Disease: A Review
- K. Aditya Shastry
- H. A. Sanjay
Multimedia Tools and Applications (2023)
Quick links
- Explore articles by subject
- Guide to authors
- Editorial policies
Sign up for the Nature Briefing newsletter — what matters in science, free to your inbox daily.


An official website of the United States government
The .gov means it’s official. Federal government websites often end in .gov or .mil. Before sharing sensitive information, make sure you’re on a federal government site.
The site is secure. The https:// ensures that you are connecting to the official website and that any information you provide is encrypted and transmitted securely.
- Publications
- Account settings
Preview improvements coming to the PMC website in October 2024. Learn More or Try it out now .
- Advanced Search
- Journal List
- Cold Spring Harb Perspect Biol
- v.7(9); 2015 Sep
Perspectives on Human Variation through the Lens of Diversity and Race
Human populations, however defined, differ in the distribution and frequency of traits they display and diseases to which individuals are susceptible. These need to be understood with respect to three recent advances. First, these differences are multicausal and a result of not only genetic but also epigenetic and environmental factors. Second, the actions of genes, although crucial, turn out to be quite dynamic and modifiable, which contrasts with the classical view that they are inflexible machines. Third, the diverse human populations across the globe have spent too little time apart from our common origin 50,000 years ago to have developed many individually adapted traits. Human trait and disease differences by continental ancestry are thus as much the result of nongenetic as genetic forces.
Humans across the globe display variation in numerous different traits. But these differences are caused by both genetic and nongenetic factors and do not define distinct “races” in biological terms.
Half-jokingly, Gwen Ifill, the noted American journalist and newscaster, told the Smithsonian audience, “In no universe is President Obama white!” ( Smithsonian’s National Museum of Natural History 2013 ). Her comment came in response to my genetic argument that given the President’s white American mother and Kenyan father, he could just as well be called “white” as “black.”
This friendly exchange exposed the essential conundrum surrounding the contemporary meaning of labels, classification, and the notion of race. We humans have, since time immemorial, sorted and classified each other into numerous categories based on language, culture, and appearance ( en.wikipedia.org/wiki/Race_(human_classification) ; Blumenbach 1775 ). Irrespective of how these groupings were decided on or justified, such classification has been a cultural exercise: the basis for self-identification through the identification of others. Genetics, being a modern science, has come to this scene much later. Genetics has much to say about the recent and remote history of our species and our individual ancestries, as well as the potential to support or refute our existing classifications. What genetics says about our history cannot be wished away. At the same time, our cultures have a strong voice in how we view ourselves and view others. This cultural view cannot be wished away either. President Obama’s self-identification as “black” is not based on his personal gene accounting but rather a nonchoice given American social convention (the “one-drop” rule) and his personal history. That was Ifill’s point. What we, and others, call us depends on both our genes and our society. I am a Bengali-American, the duality and hyphen being equally important to my identity over and above my genes.
So, what are we to think of the existence of human “races” in this “genomic age” and why is there still so much controversy? ( Koenig et al. 2008 ). By any account, we cannot ignore the idea of human races. It is in daily common use, a basis of self-identification and for many a key to their social identity. Without quibbling about word usage and specific meaning, race is also the basis for governmental statistical accounting and political action. Although the word race finds daily use in the United States, and, increasingly in Europe, similar controversies surround other classifications of humans, such as caste or tribe, despite the diverse origins of each term ( Thapar 2014 ). The most controversial aspect of such classifications, in my opinion, is not whether they are biological or not, but rather the imputation of wholesale traits 1 and attributes to these groups so defined. Almost without exception, the characteristics displayed by one’s own group are deemed positive and implicitly valued, whereas those of other groups are deemed negative and are undervalued. The group defining and possessing the valued features is also invariably that group that is culturally dominant and politically powerful.
Genetics has always been an ideal fuel for this fodder. It is a science that examines the biological basis for trait differences, and has recently allowed us to make tremendous strides in our understanding of human disease (e.g., why some individuals have muscular dystrophy and others do not) and differences (e.g., why some individuals can digest milk and others cannot). The argument goes that if groups can differ in traits such as lactose intolerance (most Europeans and some Africans are tolerant, whereas others of the world are not) and malarial susceptibility (many Africans, Middle Easterners, South Asians, and East Asians have some protection, whereas the rest of the world does not) owing to specific gene differences, they, in all probability, also genetically differ in a host of health-related traits. But, why should this principle be restricted to health-related features? Some have argued, why not genetic differences in intellectual ability, industriousness, the facility for democratic institutions, or aggressive behavior ( Wade 2014 )? In the past 100 years of genetics, many in the field have advertently and inadvertently engaged in considerable speculation as to these last possibilities, the biological underpinnings of any difference. But what is the evidence that these metafeatures are genetic? I know of no science that can prove the genetic underpinnings of these broad social differences. In contrast, I know of plenty of evidence that argues against it being the case ( Chakravarti 2010 ). As the physicist Neil deGrasse Tyson recently quipped: “You get to say the world is flat because we live in a country that guarantees free speech, but it is not a country that guarantees that anything you say is correct” ( deGrasse Tyson 2014 ).
Over the next several pages, I would like to tell you what we know of human diversity and how we came to be who we are today: in short, our singular genetic heritage and history ( Fig. 1 ). I will outline what we know today about genetic differences across contemporary human populations and how these differences can sometimes account for the human trait differences we observe. I will not opine on whether race exists or not; it does so in a very real sense. Instead, I will discuss what modern genetics says or does not say about the genetic meaning of race. Such work, together with increasing knowledge of how genes function at the molecular level, is giving us new insight into how genes influence our traits. But we remain vastly ignorant; so wild speculations about the genetic nature of many human attributes reside beyond the realm of today’s science. Importantly, the notion of the gene, in the minds of most—many geneticists included—is one of an inflexible machine with deterministic outcomes. As the science advances, we increasingly find that the effects of genes are highly modifiable, dynamic, and subject to external influences ( Chakravarti 2010 ). Indeed, why only genes? Even their ultimate products, structures such as our brain, are highly modifiable and dynamic ( Kays et al. 2012 ). Overlaid on top of all of this are the vast demographic changes human populations are increasingly experiencing, driven by increased communication and movement, and the shedding of past cultural divisions. These changes profoundly affect the distributions of genes across humanity and the melding of what were once population attributes. The science of genetics will be crucial to understanding how human history unfolds, and I predict that most of our current prejudices will turn out to have no biological basis.

The long trek of our ancestors from the beginnings in Africa ∼150,000 years ago to their emergence out of Africa about 50,000 years ago to colonize the Levant, Europe, Asia, Australia, Americas, and eventually, Oceania. (From Gluckman et al. 2009 [Fig. 6.6, p. 142]; reprinted, with express permission, from the authors in conjunction with Oxford University Press © 2009.)
WHY ARE WE NOT ALL THE SAME?
There would be nothing to argue about if humans were not different from one another. Of course, there are the rare exceptions of identical twins, but even though they have identical genomes they can on occasion show different traits. This perceptible difference within a collection of similar things extends across all of nature. Science is possible only because these differences exist and is driven by our continual quest to find out how and why they arise. Genetics is a young science, its 100-year history arising from the quest to find how and why biological differences arise and how they are maintained. The “how” was first answered by Gregor Mendel’s experiments with peas and the “why” by Charles Darwin’s wondrous voyage to the Galapagos ( Provine 1971 ). We persist in continuing to answer these questions in ever more detail because our current understanding, despite being solid, is very incomplete. We are acutely aware of what is not true but often on shaky ground about what is.
We differ in traits because of the biological processes that produce us. Some of this is genetically encoded and some of it is environmentally induced or modified. Each of us develops via a genetic program that is encoded by the sequence of A, C, T, and G bases in the DNA that makes up our genomes. Each of us inherits two genomes, one from each of our parents. This genetic program defines each of us uniquely and is more similar between any two members of the same species than between members of two related species. Thus, human genomes are more similar to one another than any one of our genomes to that of our great ape relative the chimpanzee. The differences do not stop there. Each of us is acutely aware of individual-to-individual differences between humans and the greater similarity between any one of us and our family members. Genetics provides a singular explanation for both observations. The closer the relationship between two persons the more similar their genomes and the greater the similarity of their traits. The dissimilarities between our genomes are owing to constantly arising mutations in our DNA. The majority of these are never transmitted to subsequent generations (they are lost), but a minority persists over time and across the generations. These surviving variations are the currency of modern genetics. Two individuals are related by virtue of sharing one or more common ancestors from whom they have inherited a small segment of their genome. The more remote the common ancestor(s), the less the fraction of the genome shared. Thus, we share 1/2 of our genome with each of our parents and siblings, 1/4 with each of our four grandparents, 1/8 with each of our first cousins, and so on ( gcbias.org/2013/12/02/how-many-genomic-blocks-do-you-share-with-a-cousin ).
The genetic mutations that persist among us, called genetic variants or polymorphisms, can occur at various frequencies within a population; some are rare and unique to a family, whereas others are common and have spread throughout humanity. These genetic variants can be assessed to evaluate how different two individuals are, and therefore figure out their relationship. The first human genetic variation identified was the ABO blood-group system in 1900, which was used almost immediately for assessing close relationships such as paternity. Today, technological advancements allow us to examine the entire genome in exquisite detail and to identify essentially all of such genetic differences. Typically, when one compares two copies of the human genome, say the maternal and paternal copies in any one of us, we find one of every 1000 bases to be different. Because the human genome is three billion bases long, that represents three million differences. Like the Hubble telescope that has allowed us to see deeper into space and time, new genomic technologies can identify all of these differences today and allow us to detect ever more remote relationships, and ancestries to 100,000 years before present or more, and do so on a global scale. The consequent stories of what these similarities and differences mean for similarities and differences in human traits and diseases are only in their infancy.
THE BIOLOGICAL BASIS FOR VARIATION IN HUMAN TRAITS AND DISEASES
Friar Gregor Mendel was the first geneticist ( Orel 1996 ). He was deeply interested in the question of how differences in plant characteristics arise and how they are propagated. His now famous experiments, using simple observable traits (plant height, seed shape, flower color) of the pea plant, allowed him to infer that trait variants were owing to differences in separate “factors,” that these factors existed as pairs in individuals, and that one of each was transmitted to each offspring, randomly and independently of other factors. Mendel’s factors are today’s genes. A fact not appreciated is that Mendel performed many other similar experiments with other traits and other plants and failed to uncover similarly clarifying principles ( Orel 1996 ). This is not to say his rules of inheritance governing genes are incorrect; these apply to all genes. Rather, some genes have overwhelming effects on a trait and so the trait inheritance patterns are simple, the so-called Mendelian traits, such as those of plant height, seed shape, and flower color in the pea plant. Other genes, however, exert their effects in concert with numerous other genes, none of which overwhelm the others and thus have more complex patterns of inheritance. The fact is that Mendelian inheritance of “traits” is very much the exception, not the rule ( Chakravarti 2010 ). The failure to understand this key feature led to considerable and bitter controversy in the early genetics literature (the Mendelian-biometrical debate) when some held that metrical traits such as height were not inherited but rather their variation arose solely from environmental differences ( Provine 1971 ).
Eighty years hence, we are considerably more informed as to the nature of non-Mendelian inheritance. Much of the science of genetics has advanced from the experimental use of Mendelian inheritance to uncover its biological and molecular underpinnings. We also know that although some traits arise from the actions of two or at most a few genes, the vast majority of traits are genetically complex arising from the actions and interactions of numerous, hundreds or even thousands of genes. We know this because contemporary genetics allows us to map the locations of the individual genes contributing to a trait and, for most, a role for hundreds of genes has been revealed ( Lango Allen et al. 2010 ). We also know that many more unmapped genes exist and that the individual-to-individual variation in a trait also involves the contributions from other domains, namely, environmental and epigenetic factors. The term environmental as used in genetics is both broad and nonspecific, and can include everything from lifestyle (diet, exercise) to ecological (weather, altitude) to social (income, education, health-care access) and cultural (diet, belief systems) factors. The term epigenetic is also broad and includes a whole host of cellular processes that can direct the actions of genes without being dependent on the sequence of a specific genome. There is also increasing evidence that cellular (genetic) outcomes are not deterministic but inherently dynamic and stochastic. Genes do not determine only one outcome, but a range of possible ones. Finally, consider that biological effects cannot be arbitrary but are both canalized (restricted to certain possibilities) and built to preserve homeostasis (physiological regulation maintaining more or less constant internal conditions). There is one more arbiter; evolution decides which of the many genetic changes that occur within our genomes will be retained and which ones will be culled depending on whether the change is beneficial or not.
The variation in any trait, or for that matter disease susceptibility in any species including our own, is the result of many factors (genetic, epigenetic, environmental) each of which can be further divided into multiple subfactors. It is no surprise that traits can be inherited in a complex manner because beyond genes, whose inheritance patterns we understand, the epigenetic and environmental factors can also be “inherited,” albeit according to rules still not understood ( Cavalli-Sforza and Feldman 1981 ; Jirtle and Skinner 2007 ). It is then also unsurprising that Mendelian inheritance of traits is rare, because these represent the unusual singular effects of one gene that overwhelms the extant nongenetic variation.
Geneticists have long been interested in the precise genetic architecture of “complex traits.” The first step is assessing a trait’s genetic component. The top-down or classical approach has been to compare traits among relatives, because we have long known how much genetic information relatives share (e.g., 50% between siblings) without knowing individual genes. This allowed us to estimate the proportion of variation that is genetic, a proportion called heritability. The concept of heritability has been of great practical utility in plant and animal breeding, as a guide for choosing which strains to develop for improvement of yield and performance-related traits. The heritability of numerous human traits and diseases has also been measured, often repeatedly. Although some traits have high heritability, like height (>80%), the overwhelming majority has low to moderate heritability (30%–50%) ( Vinkhuyzen et al. 2013 ). The specific identification of the genes that explain this heritability, by contemporary bottom-up approaches in which the entire genome is systematically investigated, has been notoriously difficult, however ( Lango Allen et al. 2010 ; Vinkhuyzen et al. 2013 ). This is sometimes referred to as the “missing heritability” problem.
There are many reasons for this apparent failure. First, our study samples are yet too small and not diverse enough. Second, our technological approaches are insufficient to recognize the vast network of gene interactions that may be principally important. Third, heritability estimates exaggerate the effect of genes because most studies cannot distinguish genetic from social or cultural sharing; family members share much more than genes (social, cultural, and dietary factors). Fourth, heritability is a relative measure of genetic versus nongenetic contributions. Thus, simply increasing or decreasing the environmental part of the variation can alter the apparent role of the genetic part. Well-known examples, such as the height increase seen from improved nutrition in the absence of any genetic change or the dietary treatment of phenylketonuria from birth to prevent intellectual disability, show that the actions of genes can be mitigated by nongenetic interventions.
WHY GENES TELL STORIES
Almost every human gene, when its genomic sequence is compared across individuals, shows variation. Each such sequence is a palimpsest, recording all changes from mutations that have survived until today. Some of these changes are unique to particular individuals, perhaps even one, whereas others are present in many of us. Because all humans belong to a single family tree—on average any two of us share 99.9% of our genomes—the fraction of sequence difference between any two genomes indicate how far back in time they had a common ancestor. Different segments of the genome are shared with different common ancestors; so the fraction shared or different between two genomes varies along its length. Consequently, examining the entire genome, as we can do today, is more informative than studying only one bit, such as the maternally inherited mitochondrial genome or the paternally inherited Y chromosome. The latter are informative nevertheless because they allow us to make inferences about our maternal and paternal lineages, respectively. Because all genetic changes accumulate over time, our genomes thus provide a history of how we, as individuals and as a species, came to be. Today we can compare individual genomes to infer our relationships, how far back in time we shared one or more common ancestors, and with increasing precision because of limited mobility of our ancestors, where our forebears were geographically located.
Genomic technologies, genomic sequencing in particular, have opened the door to recovering our individual and collective genetic histories, and, therefore, in concert with other sources of information, to uncovering details of our prehistory. In this sense, genes tell compelling stories about each of us as well as our shared humanity. This is a truly remarkable scientific and social achievement. There are many aspects of these stories that are uncertain and will require revision in the future. Nevertheless, some compelling and surprising truths have emerged. The most important of these is that contemporary humans are a remarkably young species and we all belong to a single family tree that arose from common ancestors a little more than 150,000 years ago ( Pääbo 2014 ). Modern humans came to be in the last few minutes of the last hour of the last day if all 14.5 billion years of cosmic evolution were compressed into one year. If we were the common bacterium Escherichia coli , then this would correspond to a mere 3 months of our life. The story of human diversity, why we look as diverse as we seem to, needs to be told with this truth in mind.
WHAT IS RACE?
The Oxford English dictionary defines human races as the “major divisions of humankind, having distinct physical characteristics,” and also as a “group of people sharing the same culture, history, language, etc.” Biologists have had a more specific definition of race, one not conjured with human diversity in mind. The evolutionary biologist Ernst Mayr wrote that a race is “an aggregate of phenotypically similar populations of a species inhabiting a geographic subdivision of the range of that species and differing taxonomically from other populations of that species” ( Mayr 2002 ). This definition has more to do with biogeography and taxonomy. However, there is an implicit assumption of both transmission and permanence of such taxonomy, and biologists impute the existence of some fundamental genetic and evolutionary difference between groups termed races. If one believes in evolution and modern genetics, and a common tree of life, the conclusion is inescapable that some members of a single species will be more different than others; additionally, close relatives of each of these members will be more similar to their closer rather than their more distant kin. It is unsurprising that this is true for humans and that our many attributes, including physical features, show this pattern. The precise pattern of sharing is a result of our specific evolutionary history and these differences are written in our genes and propagated through them. The construction and existence of human races in this regard, quite apart from social and cultural meanings, would not per se be controversial. It is controversial today because, over the past few centuries, both experts and nonexperts alike have brought in new and corrupted meaning that is not inherent in the biological concept. Discussions on human race, and caste, are difficult and incendiary today because their subtext is that human genetic differences are not neutral but either advantageous or disadvantageous and, tragically for human history, the corollary is that some groups have mostly advantageous attributes, whereas other groups have largely disadvantageous ones ( Herrnstein and Murray 1994 ; Koenig et al. 2008 ; Wade 2014 ). Genetics has provided a second, more pernicious, corollary. Because some of these traits might be “genetic,” these differences are transmitted at conception and so are biologically permanent ( Herrnstein and Murray 1994 ; Wade 2014 ). The implication is that some groups have a genetic advantage. There have never been any empirical data to support these claims, and, moreover, the survival of “diverse” human groups is prima facie evidence of each of our groups’ evolutionary success ( Fraser 1995 ).
We humans must have always named and classified each other as we came across our evolutionary kin. However, the rise and expansion of the modern concept of human races had to wait for the conquests by colonial powers that brought Europeans into direct contact with many groups who were different with respect to language, culture, and even physical features. It should be remembered that this was always an asymmetrical and unequal rendezvous, favoring the colonizer and disfavoring the colonized. The notion of human races arose in this background from studies of comparative anatomy of human skulls by the German physician and naturalist Johann Blumenbach in the 18th and 19th centuries ( Blumenbach 1775 ). He classified these skulls, and thereby humanity, into five major classes—Caucasian, Mongolian, Malayan, Ethiopian, and American—and began the horrid practice of providing color aliases (white, yellow, brown, black, and red). He correctly concluded that “individual Africans differ as much, or even more, from other individual Africans as Europeans differ from Europeans.” Blumenbach, like his contemporaries, believed in the “degeneration hypothesis,” which is that humans were originally “Caucasian” and that other races were the outcomes of environmental degeneration (e.g., through exposure to sunlight). Despite the cultural biases he began with, Blumenbach was far more generous than his contemporaries with his view that Africans were not intellectually lesser than their European counterparts ( Blumenbach 1775 ). Subsequently, despite other investigators classifying humans into anywhere from two to 63 races, no less an authority than Charles Darwin opined that “it is hardly possible to discover clear distinctive characters” between human races, because they “graduate into each other” ( Darwin 1871 ).
The political and economic rise of Europe, and then the United States, in the 19th and 20th centuries, fed many bogus ideas into what evolved as “scientific racism” ( Fredrickson 2002 ). There were parallel developments dealing with caste differences in India, although this is a much older classification. These studies had in common the examination of selected traits and attributes, deemed to be hereditary, in turn justifying the conclusion that there was a well-defined value hierarchy inherent in our species, with some groups much better endowed than others. The new emerging concepts of genetics added a new dimension; heritability assured that both the well- and less-well endowed continued to remain so. These beliefs—and they are largely beliefs of the perpetrators because so much of their data has been subsequently shown to be selectively used, manipulated, and outright fraudulent—led to a long period of eugenics both in the United States and Europe and the subsequent rise of the concept of inherent group superiority. This had disastrous consequences for Jews, Gypsies, intellectuals, nonconformists, and the mentally ill, among others, and biased immigration policies in many countries, including the United States. Unfortunately, geneticists had no small role to play in this crime of historical proportions ( Witkowski and Inglis 2008 ). As a number of the authors in this collection describe, there is still a continuing tendency to conflate all manner of group differences with gene differences ( Herrnstein and Murray 1994 ; Cooper 2013 ; Duster 2014 ; Wade 2014 ).
In the next section, I will turn to a modern accounting of human population variation and what it may say or not say about human races. Irrespective of that answer, one aspect is certain. Modern genetics research does not support the contention that one group or another has all of the positive traits, that we understand the genetics of complex traits sufficiently well enough to know that group differences in traits mean majorly group differences in genes, or that traits with a genetic component have fixed, inviolate, permanent, and unmodifiable effects. Even if we were to defend the idea of human continental ancestry or race it would be impossible to defend the assertion that they are inherently unequal. The remarkable feature of human evolution and adaptation is the widespread commonality of highly advantageous features (speech, cognition, culture) throughout humanity and the less frequent evolutionary innovations that occurred locally (pigmentation).
A BRIEF SYNTHESIS OF RECENT HUMAN EVOLUTION
The evolution of hominids leading to Homo erectus , 1.5 to 2.5 million years ago, and then Homo sapiens in Africa, is now well established. Although H. erectus existed outside Africa, the evidence is very clear that we are all descendants of groups from the African continent. H. sapiens first appeared there no earlier than ∼300,000 years ago ( Klein 1989 ). The subsequent history, evident in only fragmentary form through fossil remains, is where genetics has been indispensable ( Cavalli-Sforza et al. 1994 ; Pääbo 2014 ).
The widespread discovery of gene variation in the 1960s immediately prompted studies to assess their relative relationships across human groups. The first study reconstructing human evolution using data from living groups was by Cavalli-Sforza and Edwards in 1964 ( Edwards and Cavalli-Sforza 1964 ). This landmark study produced a “tree” in which extant populations arose through independent evolution by splitting from a common ancestral group that also produced a sister group, and so on. This study yielded two major findings beyond the specific relationships between groups. First, geographic proximity reflected greater genetic similarity across all groups, with the largest difference being between African and Australian Aboriginal samples. This suggested that human colonization occurred through successive and serial migrations. Second, anthropometric measures and skin color showed a very different set of relationships, for example, a close association between African and Australian Aborigines, unrelated to geography but dependent on climate ( Cavalli-Sforza and Edwards 1964 ). Another landmark study by Richard Lewontin in 1972 went on to show that the majority of human genetic variation, on average 85%, existed within any group and that intergroup differences were relatively minor, with the largest being between African and non-African groups ( Lewontin 1972 ). These were not isolated controversial studies but rather the beginning of an onslaught of investigations, using successively larger and larger numbers of genes and humans, which have produced a single, consistent genetic narrative of human history ( Cavalli-Sforza et al. 1994 ).
All early studies of human evolution compared the features and relationships of populations not individuals. In other words, these studies compared the relationships between frequencies of gene variants and not the genomes of individuals. This distinction is critical because past studies depended on the definition of a population. Is it defined by language, culture, geography, physical appearance, caste, or “race?” The definitions, of course, could skew the results one way or another. Of course, populations defined by known specific differences can be different at the genetic level. This is why Allan Wilson’s 1987 study of individuals and their mitochondrial genomes is a significant departure from the past ( Cann et al. 1987 ). His research accomplished four major goals. First, they studied individual genomes and not ensemble frequencies; second, they clearly showed that the human evolutionary tree had two major branches, one composed of African mitochondrial genomes only and the other comprising all humans including Africans; third, except for Africans, all other individuals from the same population had multiple origins; and fourth, they dated the common mitochondrial ancestor of all humans to be less than 200,000 years ago.
There have since been many genetic studies using ever-increasing types and numbers of genetic variants and culminating in contemporary studies involving whole genome sequencing of diverse collections of humans. The essential conclusions of the findings described above have now stood the test of time. The single story of human evolution in the last 150,000 years is that all of us today are the descendants of early H. sapiens in Africa. A small group of these ancestors migrated “out of Africa” about 50,000 years ago and have since colonized the rest of our globe with each new group serially colonizing new unexplored geography. This is how we came into the Levant (Middle East) and then to both Europe and Asia and its subcontinent, and beyond into New Guinea and Australia. This is also how we went to remote parts of Siberia and came to colonize the New World about 15,000 years ago, which was until then hominid free. Our latest forays, only 2000 years ago, have been into Oceania ( Fig. 1 ). These journeys provide the explanation for the pattern of quantitative differences that Cavalli-Sforza, Lewontin, and Wilson first brought attention to. Yes, there are differences in genetic variation at the continental level and one may refer to them as races. But why are continents the arbiter? If humans have had this single continuous journey disobeying continental residence—and as evidence we have the continuous distribution of genetic variation across the globe, not discrete boundaries like political borders—where do we divide humanity and why? ( Weiss and Lambert 2014 ). All humans, without exception, are one species that has only very recently dispersed, with each population being more related to its proximal geographic neighbors. If we do look, behave, and have features that distinguish us markedly from one another, then these are differences that have arisen and amplified only over the last 50,000 years (2000 generations) and, quantitatively, are very small—only one part in 1000 bases. As a comparison, consider that chimpanzees and humans diverged from a common ancestor more than five million years ago (200,000 generations).
Of course, there are many more details to fill in and we wish to have far greater resolution of the history we already know. In this collection, scholars of genetic variation and evolution in the major geographic regions of the world have outlined both known and more recent genetic studies ( Gomez et al. 2014 ; Majumder and Basu 2014 ; Ruiz Linares 2014 ; Veeramah and Novembre 2014 ). Their research, and the complementary works of others, allows us to make three major novel inferences. First, human populations were not always large and, probably, were almost always small. Second, the current abundance of a group is not a reflection of its past size. Third, human populations are seldom homogeneous and are highly admixed. 2
We academics and nonacademics alike like to associate the genome only with its biological properties. However, our genomes and genetic variation between them and peoples are also the result of who lives, who dies, and who leaves behind how many offspring (i.e., demography). In other words, our genomes carry the record of both biology and demography, although teasing these apart is neither trivial nor easily corroborated by independent sources of information. In fact, contemporary whole genome genetic variation data emphasize the greater imprint of demography than biology in our genomes. As mentioned above, the chief conclusion of many genetic studies is that only a small group emerged from Africa to colonize the world and each successive colonization involved small numbers of founders as well ( Gutenkunst et al. 2009 ), sometimes in the hundreds. This speaks to the limited amount of variation each founding group carried (which was also a subset of that in its parent group) and the constant threat of extinction to such a small band. Our eventual success is often chalked up to crucial adaptations; however, it is not implausible that there were many attempts, many catastrophes, and we are simply the lucky survivors. Of course, long-term survival came from population expansions, an intrinsic feature of human, and all other, evolutionary success. As other authors of this collection have documented, human population sizes rose within Africa at the time of our early ancestors, and then outside Africa as their descendants spread across the globe. These population increases must have depended on many environmental and chance factors as well as the inclusion of new arrivals (immigration). These factors are neither stereotypical nor orderly, once again emphasizing the strong effects of chance. A group populous today was not necessarily originally so; on the contrary, they may have been ever so close to extinction. Finally, although in the minds of many genetics is associated with homogeneity, human evolution is nothing else but a story of admixture and heterogeneity. There is now ample evidence in the genetic structures of the peoples of Africa, Asia, Europe, and the Americas that all extant humans are admixed (this collection). The new data are only now revealing whom we were admixed with, whether such admixture was common or rare, and when it occurred. Indeed, it is this story of mixing that is so at odds with the classical view of human group identity ( Reich et al. 2009 ). We tend to think of admixture as a feature of modern times and, as has frequently happened, in terms of subjugation of one group by another as has occurred many times across human history and geography. However, this must have occurred even in the remote past; as human population density increased, there must have been a greater frequency of encounters with others and thus opportunities for mixing. The recent evidence that many humans carry genomic segments that can be traced back to Neanderthals and Denisovans (a second archaic human group) is evidence of such genetic exchange even 30,000 or more years ago with then-existing archaic human groups ( Pääbo 2014 ).
The history that I have outlined above has implications for the natural selection and adaptation that have surely shaped our genomes. First, the major adaptive events that led to the emergence of H. sapiens are not the subject of debate. For matters of race, it does not even matter what happened to our young species in all of the last 150,000 years, or the emergence of modern humans while in Africa. What does matter are the adaptations in the past 50,000 years that have led to our spread across all continents and the genetic differences between us since then. This span of 2000 generations is long enough for specific adaptations to have occurred but not for very many such adaptations. The reasons, albeit technical, essentially depend on the following argument. Every adaptation occurs through some individuals possessing a beneficial mutation that, while bringing them an advantage (larger numbers of surviving offspring), is a relative disadvantage to those who do not carry that beneficial variant. This is a precarious gift because early in the evolution of this mutation most individuals are at a disadvantage and even those with an advantage might not realize their benefit. The English geneticist J.B.S. Haldane argued that this cost of selection was high enough that many beneficial mutations cannot arise at the same time ( Haldane 1957 ). Of course, adaptations do occur as is evidenced by the striking examples of lactose persistence (selected after dairy farming arose) or skin pigmentation (selected for in response to solar radiation) ( Quintana-Murci and Barreiro 2010 ). To me, even more striking is the repeated birth of the sickle cell mutation at the same DNA site in Africa as a protective response to malaria ( Wainscoat et al. 1983 ). Nevertheless, these examples of adaptation are rare and there are likely only a handful of such examples in genomes across humanity ( Hernandez et al. 2011 ). Positive selection and adaptations must have occurred, but their mechanisms likely are not through single genes but across many genes affecting the same trait, as is the case for height ( Turchin et al. 2012 ). If that is the common scenario, then even strong selection on one gene among the many that affect a trait is going to be trivial unless we are speaking of very long evolutionary times or adaptive changes that occurred in our shared history.
The thesis that human groups substantially differ in most traits that are deeply rooted in simple genetics, and the result of recent adaptation, is fanciful. Human groups do differ from one another in many ways and the reasons are more likely to be nongenetic than genetic. Of those that are genetic, their composition is likely owing to many genes (hundreds to thousands), adaptations at many of them unlikely to be sustainable by known genetic mechanisms in the time frame during which human differences must have accumulated. More than 45 years ago, Motoo Kimura contended that, broadly, most of molecular evolution is deleterious and doomed to extinction; of those that do survive, the vast majority are selectively neutral ( Kimura 1968 ). Recent data suggest this to be amply true. A benefit of this theory is that it means that the vast majority of changes in our genomes occur at a constant rate and provide an excellent “molecular clock” to date specific events in our common and unique histories.
THE NEXT PHASE OF HUMAN GENETIC DIVERSITY STUDIES
Human genetic diversity is dynamic and its patterns have changed substantially over time and will change in the future. Each of us can trace our genomes back to Africa and the subsequent journey across 50,000 years with intermixing with many other peoples. We live, however, in very different times. There are increasing rates of meeting and mixing, including groups that may have been relatively isolated for a few thousand generations. There are also rapid cultural and social changes that make neither gene nor cultural isolation possible. All of this implies even greater admixture than ever before. Consequently, I suspect the study of individuals and their genomes will increase at the expense of studying populations. It is remarkable how many individuals choose to study their genomes purely to decipher their ancestry ( www.23andme.com ). These individual genomes will surely uncover their individual histories but also begin to add detail to our common genetic history ( 1000 Genomes Consortium 2012 ).
I suspect that the focus on race, caste, or tribe that we still see today will erode simply because fewer and fewer members of any group will have its hallmark features. What does continental ancestry mean when one is from more than one? It might survive, I suppose; after all, how many fans of Manchester United across the globe have ever even been to Manchester? For a while, our ability to decipher individual histories might be useful to test how strict or porous the concept of a “population” is. Genetics and evolutionary biology have held as fundamental the concept that a population is a real, stable genetic unit, a property that is discrete and survivable. The reality is that most populations are dynamic and fluid, neither real nor stable.
Human evolution has always been studied with respect to such populations defined by language, geography, or cultural and physical features. Consider instead what we could decipher if we could sample a million humans (say), without regard to who they were, across a virtual grid across the world; this would correspond to sampling one person every 57 square miles (∼7.5 miles × 7.5 miles) across the land surface. (This grid sampling idea was mentioned to me by the late Allan Wilson sometime around 1988.) Assume as well that we would sequence their maternal and paternal genomes and ask them questions, such as where they were born, where their parents were born, which language(s) they spoke, and what group affiliation(s) they had. We could then specifically uncover not only all of the features of human evolution we know today, and revise them to greater accuracy, but also test whether any or all of the features we use for human classification are supported by their genes. These types of global surveys of diversity have been performed for other species and may provide the first objective description of ours, bereft of race and other labels. This does not vitiate any social or cultural ways of defining humans, but at least one can no longer claim a genetic basis for all group differences.
RACE-BASED AND INDIVIDUALIZED MEDICINE
One of the major contributions of genetics to medicine has been, beyond the identification of disease pathophysiology, the recognition that each disease is multicausal and that patients with a single disease label may have conditions that arise from distinct molecular causes. This is well recognized for single gene disorders such as muscular dystrophy or even broad categories like prelingual deafness. Distinct molecular etiologies may require distinct therapies and management methods simply because physicians are trying to ameliorate different pathologies. These ideas have led to the concept of individualized medicine, that is, the tailoring of care to each patient depending on their genetic makeup and individual health circumstances ( Childs 1999 ). This conceptual basis for care is a fundamental change in medicine that has long relied on the idea of a typological patient. Medicine’s goal is now individualized care for the common chronic human diseases, a far more challenging task because the genetic and molecular bases for most chronic diseases remain unknown. We are making great progress but we have a long journey ahead before we understand the genetic, epigenetic, and environmental contributions to these disorders and which of these three may be the best route to intervention. That, of course, should not prevent us from individualizing care as best as we can with the knowledge we already have.
One of the major areas for individualized medicine is cancer therapy in which molecular diagnosis of germline mutations has been prevalent for more than two decades, and the genetic profile of the tumor has directed aspects of treatment. Increasingly, genome sequencing is being used to profile tumors broadly to direct treatment ( Vogelstein et al. 2013 ). Individualizing risks to specific cancer subtypes and tumor profiling are expected to become routine aspects of cancer treatment particularly because personalized immune-modulation therapy is also on the horizon ( Pardoll 2012 ). It is interesting to reflect that so much progress has been made in cancer treatment with greater discussion of personalized medicine rather than through the lens of race, despite cancer epidemiology data that show differences by continental ancestry. Differences in cancer incidence and prevalence by ancestry or ethnicity or community are well known. In fact, significant differences by geography, in the United States down to the county level, are also well known, suggesting major environmental etiologies as well. These differences, which lie beyond genes, need to be addressed simultaneously. The tussle lies in which genes and environments will we emphasize when both are responsible?
There is, of course, a long history of the study of variation in the incidence and prevalence of any disease by race, continental ancestry, and ethnicity. There is no doubt that many disorders show persistent and consistent differences and lead to great health disparities around the world ( Murray et al. 2013 ). Genes do contribute to some part of it, probably no more than half, based on heritability studies, but environmental, epigenetic, and chance effects contribute significantly as well. Thus, equating all differences to genes is neither correct nor wise. As the essays by Richard Cooper and Troy Duster eloquently argue, the nongenetic factors in human disease, equally if not more importantly, affect our genetic biology in fundamental ways, and in many cases merit direct interventions that can lead to reductions in disease prevalence. Treatment of elevated blood pressure to prevent hypertension and its associated damage to the heart and kidney is a cogent example. In the United States, African Americans have elevated rates of hypertension and its sequelae as compared with those with European ancestry. This consistent finding has led to the myth that Africans have a higher genetic predisposition to hypertension, a fact clearly refuted by studies of many African communities whose blood pressures are lower than those of many European communities ( Cooper et al. 2005 ). Moreover, recent genetic investigations in very large samples clearly show that blood pressure susceptibility variants detected in European ancestry subjects are also susceptibility variants for African, Asian, and South Asian subjects ( Ehret et al. 2011 ). How could it not be so? Blood pressure regulation is a crucial human physiological trait under homeostasis, probably modulated by hundreds of genes, genetic variation of which was probably chosen in the early days of human evolution. It is not surprising that all humans likely share such variants. This is not to argue that additional variants did not arise later, variants that are consequently expected to show geographic clustering, but they are expected to be fewer. In other words, a race-based approach to medicine is a poor proxy for the type of genetic understanding we need to allow advanced medical interventions, like those available for cancer. Genetics can be far more useful in identifying the molecular underpinnings of human disease and treatment differences by focusing on all of humanity and including our diversity, not ignoring it (see Lu et al. 2014 ). Population differences do exist, but genes are not the sole agent for these differences, and nongenetic factors may have greater potency ( Kahn 2013 ). An important corollary is that we should not speak of nature and nurture generally but take their respective roles on a case-by-case basis.
The challenge for understanding complex traits is thus considerable. Defining the roles of specific genes in the common chronic disorders of our time will lead to a much improved understanding of how and why a disorder develops (pathophysiology) and thus lead to improved therapies. And this progress will critically depend, I believe, on parallel progress in our understanding of how environmental and epigenetic factors impact our biology. These are the other sides of the genetic coin and there is no intellectual solution to one without the other. One of my mentors, Ching Chun (CC) Li, once remarked, “We are geneticists, not hereditarians; of course, the environment is important.” As a plant breeder in his native China he had seen both genetically selected crops wither in a drought and the fantastical bogus claims of improved yields by Trofim Lysenko. We have to get the genetics content right this time. It is a fundamental biological challenge and an exciting one. It is even more exciting to figure out how the outside (environment) affects the inside (the genetic program) and, in parallel, how and why we as a species are so much more diverse in our cultures than in our genes. Vive la différence!
ACKNOWLEDGMENTS
I am indebted to Richard Cooper, John Novembre, and Richard Sever for critical comments on this paper.
1 The word trait is used to indicate a distinguishing quality or characteristic; in its genetic flavor it means the “phenotype” or the manifesting feature of our genes (“genotype”).
2 Admixture is used here in the genetic sense, in which extant genes and individuals arise from two different ancestries, as historically occurred with the colonization of the Americas by Europeans.
Editor: Aravinda Chakravarti
Additional Perspectives on Human Variation available at www.cshperspectives.org
* Reference is also in this collection .
Want to create or adapt books like this? Learn more about how Pressbooks supports open publishing practices.
6.2 Genetic Variation
Created by: CK-12/Adapted by Christine Miller
Jumping for Joy!
The people in Figure 6.2.1 illustrate some of the great phenotypic variation displayed in modern Homo sapiens. The lighter-skinned men in the photo are Euro-American tourists in Kenya (East Africa). The darker-skinned men are native Kenyans who belong to a tribal group named the Maasai. These men come from populations on different continents on opposite sides of the globe. Their populations have unique histories, environments, and cultures. Besides differences in skin colour, the men have different hair and eye colours, facial features, and body builds. Based on such obvious physical differences, you might think that our species is characterized by a high degree of genetic variation. In fact, there is much less genetic variation in the human species than there is in many other mammalian species, including our closest relatives — the chimpanzees.
Overview of Human Genetic Variation
No two human individuals are genetically identical unless they are monozygotic (identical) twins. Between any two people, DNA differs, on average, at about one in one thousand nucleotide base pairs. We each have a total of about three billion base pairs, so any two people differ by an average of about three million base pairs. That may sound like a lot, but it’s only 0.1% of our total genetic makeup. This means that two people chosen at random are likely to be 99.9 per cent identical genetically, no matter where in the world they come from.
At an individual level, most human genetic variation is not very important biologically, because it has no apparent adaptive significance. It neither enhances nor detracts from individual fitness. Only a small percentage of DNA variations actually occur in coding regions of DNA — which are sequences that are translated into proteins — or in regulatory regions, which are sequences that control gene expression. Differences that occur in other regions of DNA have no impact on phenotype . Even variations in coding regions of DNA may or may not affect phenotype. Some DNA variations may alter the amino acid sequence of a protein, but not affect how the protein functions. Other DNA variations do not even change the amino acid sequence of the encoded protein.
At a population level, genetic variation is crucial if evolution is to occur. Genetically-based differences in fitness among individuals are the key to evolution by natural selection. Without genetic variation within populations, there can be no differential fitness by genotype, and natural selection cannot occur.
Patterns of Human Genetic Variation
Data comparing DNA sequences from around the world show that only about ten per cent of our total genetic variation occurs between people from different continents, like the American tourists and African Maasai pictured in Figure 6.1.1. The other 90 per cent of genetic variation occurs between people within continental populations, such as between North Americans or between Africans. Within any human population, many genes have two or more normal alleles that contribute to genetic differences among individuals. The case in which a gene has two or more alleles in a population at frequencies greater than one per cent is called a polymorphism . A single nucleotide polymorphism (SNP) involves variation in just one nucleotide in a DNA sequence. SNPs account for most of our genetic differences. Other types of variations (such as deletions and insertions of nucleotides in DNA sequences) account for a much smaller proportion of our overall genetic variation.
Different populations may have different allele frequencies for polymorphic genes. However, the distribution of allele frequencies in different populations around the world tends not to be discrete or distinct. Instead, the pattern is more often one of gradual geographic variations, or clines , in allele frequencies. You can see an example of a clinal distribution of allele frequencies in the map (Figure 6.2.2) below. Clinal distributions like this may be a reflection of natural selection pressures varying continuously over geographic space, or they may reflect a combination of genetic drift and gene flow of neutral alleles.
Although most genetic variation occurs within rather than between populations, certain alleles do seem to cluster in particular geographic areas. One example happens with the Duffy gene. Variations in this gene are the basis of the Duffy blood group, which is determined by the presence or absence of a red blood cell antigen, similar to the more familiar ABO blood group antigens. The genotype for having no antigen for the Duffy blood group is far higher in African populations and in people who have African ancestry than it is in non-African people, as indicated in the following table . Genes (such as the Duffy gene) may be useful as genetic markers to establish the ancestral populations of individuals.
Table 6.2.1
Population Frequencies for No Antigen in the Duffy Blood Group
The reason for the different population frequencies for the Duffy antigen appears to be natural selection. People who lack the Duffy antigen are relatively resistant to malaria , which is one of the oldest and most devastating human diseases. Malaria has been a persistent and widespread disease in sub-Saharan Africa for tens of thousands of years. DNA analyses suggest that the allele associated with lack of the Duffy antigen evolved at least twice in Africa and was strongly selected for, causing it to increase in frequency. The Duffy gene is just one of many genes that have polymorphic alleles, because one of the alleles protects against malaria. In fact, a greater number of known genetic polymorphisms may be attributed to selection because of malaria than any other single selective agent.
Factors Influencing the Level of Human Genetic Variation
The age and size of a population increases the genetic variation within that population. You would expect an older, larger population to have more genetic variation. The older a population is, the longer it has been accumulating mutations. The larger a population is, the more people there are in which mutations can occur. Anatomically modern humans evolved less than a quarter million years ago, which is a relatively short period of time for mutations to accumulate. Our population was also quite small at some point in the past, perhaps consisting of no more than ten thousand adults, which reduced genetic variation even more. These factors explain why humans are relatively homogeneous genetically as a species.
What We Can Learn From Knowledge of Human Genetic Variation
Knowledge of genetic variation can help us understand our similarities and differences, our origins, and our evolutionary past. It can also help us understand human diseases and — hopefully — find new ways to treat them.
Human Origins
The data on human genetic variation generally supports the out-of-Africa hypothesis for human origins. According to this hypothesis, the common ancestor of all modern humans evolved in Africa around 200 thousand years ago. Then, starting no later than about 60 thousand years ago, part of the African population left Africa and migrated to Europe and Asia. As the migrants spread throughout the Old World , they replaced (and/or absorbed) the populations of archaic humans they encountered.
Most studies of human genetic variation find there is greater genetic diversity in African than non-African populations. This is consistent with the older age of the African population proposed by the out-of-Africa hypothesis. In addition, most of the genetic variation in non-African populations is a subset of the variation in African populations. This is consistent with the idea that part of the African population left Africa much later and migrated to other places in the Old World.
Recent comparisons of modern human and archaic human (including Neanderthal and Denisovan ) DNA show that interbreeding occurred between their populations, but to differing degrees. The result of new DNA sequences entering a population’s gene pool through interbreeding is called admixture . There is greater admixture with archaic humans in modern European, Asian, and Oceanic populations than in modern African populations. Populations with the greatest admixture are those in Melanesia . About eight per cent of their DNA came from archaic Denisovans in East Asia.
Human Population History
Patterns of human genetic variation can be used to reconstruct population history. That history is literally recorded in our DNA. Any major population event (such as a significant reduction in population size or a high rate of migration) leaves a mark on a population’s genetic variation.
- Going through a dramatic size reduction decreases intra-population genetic variation (variation occurring within a population). As a case in point, DNA analyses suggest that there may have been drastic size reductions in the human populations that colonized the New World between 15 thousand and 20 thousand years ago. There were also size reductions in the human populations that first left Africa at least 60 thousand years ago, which helps explain the lower genetic diversity of modern non-African populations.
- A high rate of migration between populations may lead to gene flow , and this changes genetic variation in two ways. Gene flow decreases inter-population genetic variation (variation occurring between populations), while it increases intra-population variation. Gene flow — primarily between nearby populations — may contribute to the formation of clines in allele frequencies, as on the map in Figure 6.2.2.
Human Genetic Variation and Disease
An important benefit of studying human genetic variation is that we can learn more about the genetic basis of human diseases. The more we understand the causes of diseases, the more likely it is that we will be able to find effective treatments and cures for them.
Some disorders are caused by mutations in a single gene. Most of these disorders are generally rare, but some of them occur at significantly higher frequencies in certain populations. For example, Ellis-van Creveld syndrome has an unusually high frequency in Pennsylvania Amish populations, and Tay-Sachs disease has a relatively high frequency in Ashkenazi Jewish populations. Albinism is another single-gene disorder that has a variable frequency. In North America and Europe, rates of albinism are approximately 1:18,000. In Africa, in contrast, the rates range from 1:5,000 to 1:15,000. Some African populations have estimated albinism rates as high as 1:1000. The photo below (Figure 6.2.3) shows an African albino man from Mali, where there is a relatively high rate of albinism. High population-specific frequencies of single-gene disorders like these may be attributable to a variety of factors, such as small founding populations and a relative lack of gene flow.
It is likely that the majority of human diseases are caused by a complex mix of multiple genes (polygenic) and environmental factors (multifactorial). Examples of polygenic, multifactorial diseases are type II diabetes and heart disease . We do not typically think of these diseases as genetic diseases, because our genes do not predetermine whether we develop them. Our genes, however, do influence our chances of developing the diseases under certain environmental conditions. Even our chances of developing some infectious diseases are influenced by our genes. For example, a variant allele for a gene called CCR5 seems to confer resistance to infection with HIV , the virus that causes AIDS.
6.2 Summary
- No two human individuals are genetically identical (except for monozygotic twins), but the human species as a whole exhibits relatively little genetic diversity, relative to other mammalian species. Genetically, two people chosen at random are likely to be 99.9 per cent identical.
- Of the total genetic variation in humans, about 90 per cent occurs between people within continental populations. Only about 10 per cent occurs between people from different continents. Older, larger populations tend to have greater genetic variation, because there is more time and there are more people in which to accumulate mutations.
- Single nucleotide polymorphisms account for most human genetic differences. Allele frequencies for polymorphic genes generally have a clinal (rather than discrete) distribution. A minority of alleles seem to cluster in particular geographic areas, such as the allele for no antigen in the Duffy blood group. Such alleles may be useful as genetic markers to establish the ancestry of individuals.
- Knowledge of genetic variation can help us understand our similarities and differences. It can also help us reconstruct our evolutionary origins and history as a species. For example, the distribution of modern human genetic variation is consistent with the out-of-Africa hypothesis for the origin of modern humans.
- An important benefit of studying human genetic variation is learning more about the genetic basis of human diseases. This, in turn, should help us find more effective treatments and cures.
6.2 Review Questions
- Compare and contrast the significance of genetic variation at the individual and population levels.
- Describe genetic variation within and between human populations on different continents.
- Explain why allele frequencies for the Duffy gene may be used as a genetic marker for African ancestry.
- Identify factors that increase the level of genetic variation within populations.
- Discuss genetic evidence that supports the out-of-Africa hypothesis of modern human origins.
- What evidence suggests that modern humans interbred with archaic human populations after modern humans left Africa?
- How do population size reductions and gene flow impact the genetic variation of populations?
- Describe the role of genetic variation in human disease.
- Explain the reasons why variation in a DNA sequence can have no effect on the fitness of an individual.
- Explain why migration between populations decreases inter-population genetic variation. How does this relate to the development of clines in allele frequency?
- The amount of mixing of modern human DNA and archaic human DNA is an example of _________ .
6.2 Explore More
The Journey of Your Past | National Geographic, National Geographic, 2013.
Svante Pääbo: DNA clues to our inner neanderthal, TED, 2011.
Why Are Some People Albino?, Seeker, 2015.
Attributions
Figure 6.2.1
Maasai_men_and_tourists_jumping by Christopher Michel on Wikimedia Commons is used under a CC BY 2.0 (https://creativecommons.org/licenses/by/2.0/deed.en) (license.
Figure 6.2.2
Geospatial_distribution_of_SNP_rs1426654-A_allele by Basu Mallick C, Iliescu FM, Möls M, Hill S, Tamang R, Chaubey G, et al. on Wikimedia Commons is used under a CC BY 2.5 (https://creativecommons.org/licenses/by/2.5/deed.en) license.
Figure 6.2.3
Mali_Salif_Keita2_400 [cropped] by unknown from The Department of State, Washington, DC . on Wikimedia Commons is in the public domain (https://en.wikipedia.org/wiki/Public_domain).
Basu Mallick C., Iliescu, F.M., Möls, M., Hill, S., Tamang, R., Chaubey, G., et al. (2013). The light skin allele of SLC24A5 in South Asians and Europeans shares identity by descent: Figure 2. Isofrequency map illustrating the geospatial distribution of SNP rs1426654-A allele across the world. PLoS Genetics, 9(11): e1003912. doi:10.1371/journal.pgen.1003912 http://journals.plos.org/plosgenetics/article?id=10.1371/journal.pgen.1003912
HealthLinkBC. (2019, November 5). Health topics: Malaria [online article]. BC Government (gov.bc.ca). https://www.healthlinkbc.ca/health-topics/hw119119
Mayo Clinic Staff. (n.d.). Albinism [online article]. MayoClinic.org. https://www.mayoclinic.org/diseases-conditions/albinism/symptoms-causes/syc-20369184
Mayo Clinic Staff. (n.d.). Heart disease [online article]. MayoClinic.org. https://www.mayoclinic.org/diseases-conditions/heart-disease/symptoms-causes/syc-20353118
Mayo Clinic Staff. (n.d.). HIV/AIDS [online article]. MayoClinic.org. https://www.mayoclinic.org/diseases-conditions/hiv-aids/symptoms-causes/syc-20373524
Mayo Clinic Staff. (n.d.). Type 2 diabetes [online article]. MayoClinic.org. https://www.mayoclinic.org/diseases-conditions/type-2-diabetes/symptoms-causes/syc-20351193
National Geographic. (2013, March 13). The journey of your past | National Geographic. YouTube. https://www.youtube.com/watch?v=RGtaq3PiIoU&feature=youtu.be
National Institutes of Health/ National Library of Medicine. (n.d.). Genes: CCR5 gene – C-C motif chemokine receptor 5 [online article]. US Government. https://ghr.nlm.nih.gov/gene/CCR5
National Organization for Rare Disorders (NORD). (2012). Ellis Van Creveld syndrome [online article]. RareDiseases.org. https://rarediseases.org/rare-diseases/ellis-van-creveld-syndrome/
National Organization for Rare Disorders (NORD). (2017). Tay Sachs disease [online article]. RareDiseases.org. https://rarediseases.org/rare-diseases/tay-sachs-disease/
Seeker. (2015, July 25). Why are some people albino?. YouTube. https://www.youtube.com/watch?v=cHRM2S_fBOk&feature=youtu.be
TED. (2011, August 30). Svante Pääbo: DNA clues to our inner neanderthal. YouTube. https://www.youtube.com/watch?v=kU0ei9ApmsY&feature=youtu.be
Wikipedia contributors. (2020, June 18). Melanesia. In Wikipedia. https://en.wikipedia.org/w/index.php?title=Melanesia&oldid=963224885
Wikipedia contributors. (2020, June 4). Old world. In Wikipedia. https://en.wikipedia.org/w/index.php?title=Old_World&oldid=960713597
Relating to twins, derived from a single ovum, and so identical.
Deoxyribonucleic acid - the molecule carrying genetic instructions for the development, functioning, growth and reproduction of all known organisms and many viruses.
A class of biological molecule consisting of linked monomers of amino acids and which are the most versatile macromolecules in living systems and serve crucial functions in essentially all biological processes.
The set of observable characteristics of an individual resulting from the interaction of its genotype with the environment.
Amino acids are organic compounds that combine to form proteins.
The differential survival and reproduction of individuals due to differences in phenotype. It is a key mechanism of evolution, the change in the heritable traits characteristic of a population over generations.
A variant form of a given gene, meaning it is one of two or more versions of a known mutation at the same place on a chromosome. It can also refer to different sequence variations for a several-hundred base-pair or more region of the genome that codes for a protein.
A discontinuous genetic variation resulting in the occurrence of several different forms or types of individuals among the members of a single species.
A substitution of a single nucleotide that occurs at a specific position in the genome, where each variation is present at a level of 0.5% from person to person in the population.
One of the structural components, or building blocks, of DNA and RNA. A nucleotide consists of a base (one of four chemicals: adenine, thymine, guanine, and cytosine) plus a molecule of sugar and one of phosphoric acid.
A measurable gradient in a single character (or biological trait) of a species across its geographical range.
An extinct species or subspecies of archaic humans who lived in Eurasia until about 40,000 years ago. They probably went extinct due to competition with or extermination by immigrating modern humans or due to great climatic change, disease, or a combination of these factors.
An extinct species or subspecies of archaic human that ranged across Asia during the Lower and Middle Paleolithic. Denisovans are known from few remains, and, consequently, most of what is known about them comes from DNA evidence.
The presence of DNA in an individual from a distantly-related population or species, as a result of interbreeding between populations or species who have been reproductively isolated and genetically differentiated. Admixture results in the introduction of new genetic lineages into a population.
The transfer of genetic variation from one population to another. If the rate of gene flow is high enough, then two populations are considered to have equivalent allele frequencies and therefore effectively be a single population.
Human Biology Copyright © 2020 by Christine Miller is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License , except where otherwise noted.
Share This Book
If you're seeing this message, it means we're having trouble loading external resources on our website.
If you're behind a web filter, please make sure that the domains *.kastatic.org and *.kasandbox.org are unblocked.
To log in and use all the features of Khan Academy, please enable JavaScript in your browser.
Middle school biology - NGSS
Course: middle school biology - ngss > unit 7, sexual reproduction and genetic variation.
- Genetics vocabulary
- Worked examples: Punnett squares
- Genetics vocabulary and Punnett squares
- Understand: sexual reproduction and genetic variation
- Apply: genetics vocabulary
- Apply: Punnett squares
- Many sexually reproducing organisms are diploid. Diploid organisms have two sets of chromosomes in each cell. These chromosomes are organized into homologous pairs.
- In order to reproduce, diploid organisms produce gametes in the form of egg or sperm cells. These gametes are haploid. Haploid gametes have only one set of chromosomes.
- Sexual reproduction happens via fertilization. Fertilization is the fusion of gametes from two parents. Fertilization leads to new, diploid offspring.
- Sexual reproduction results in genetic variation , or genetic differences, between parents and offspring. Offspring inherit one set of chromosomes from each parent. So, an offspring has a mixture of chromosomes (and alleles) from its two parents.
- Sexual reproduction also results in genetic variation among siblings. When a parent forms a gamete, only one chromosome from each homologous pair is included at random. So, an offspring might inherit a different combination of chromosomes (and alleles) compared to its siblings.
Want to join the conversation?
- Upvote Button navigates to signup page
- Downvote Button navigates to signup page
- Flag Button navigates to signup page

Frontiers for Young Minds
- Download PDF
What Is Genetic Diversity and Why Does it Matter?

All living things on Earth contain a unique code within them, called DNA. DNA is organised into genes, similar to the way letters are organised into words. Genes give our bodies instructions on how to function. However, the exact DNA code is different even between individuals within the same species. We call this genetic diversity. Genetic diversity causes differences in the shape of bird beaks, in the flavours of tomatoes, and even in the colour of your hair! Genetic diversity is important because it gives species a better chance of survival. However, genetic diversity can be lost when populations get smaller and isolated, which decreases a species’ ability to adapt and survive. In this article, we explore the importance of genetic diversity, discuss how it is formed and maintained in wild populations, how it is lost and why that is dangerous, and what we can do to conserve it.
Why is Everything and Everyone A Little Bit Different?
Earth contains millions of different species that all look different from one another. While some species look more similar to each other than others, like lions and tigers, they will still have differences between them. Even within each species, individuals look similar to each other but they are not identical. These differences and similarities are because of many small differences between individuals’ genes . All organisms have DNA and each individual’s DNA is organised into genes. These contain the instructions to build our bodies. This is similar to the way that letters are combined to make words that then make a story. DNA can be seen as the letters, genes the words, and their instructions are the story. Small differences in DNA might change blue eyes to green, or a butterfly’s wings from black to white, like how a word can change when you replace a letter.
The combined differences in the DNA of all individuals in a species make up the genetic diversity of that species. Genetic diversity causes individuals to have different characteristics, which we can see even in our groceries. Although all tomatoes belong to the same species, the tomatoes we eat are hugely diverse, ranging from giant beefeater tomatoes to tiny cherry tomatoes. There are also hundreds of apple varieties ( Figure 1 ), that range from red to green, tart to sweet, and some apples even have pink flesh inside! Genetic diversity is what makes these types of tomatoes and apples look so different [ 1 ]. Genetic diversity is also seen in animals. For example, dogs can be large enough to pull sledges or small enough to sit nicely on your lap. All dogs are from the same species, but they look different because of genetic diversity! Though often more difficult to see, genetic diversity is also extremely important in wild animals and plants.

- Figure 1 - An example of genetic diversity in the food we eat.
- All these apples are one species. Different alleles of the genes that control their colour cause the apples to be green, yellow, red, or almost purple. Differences in the alleles that control flavour make each type taste different.
How is Genetic Diversity Generated?
Changes to an individual’s DNA are called mutations ( Figure 2 ). Mutations can arise when mistakes are made while cells are copying DNA, like making a spelling mistake when copying a word. These mutations make up a species’ genetic diversity. Over generations, more and more mistakes are made, leading to more mutations. Most mutations are either harmful or have no impact at all, but sometimes these mutations can cause changes that are helpful for a species. The individuals that have these helpful mutations might have greater chances of survival, and have more babies as a result [ 2 ]. This is adaptation . When a mother and a father have babies, the DNA of their baby is a mix of the parents’ DNA. Babies have two copies of every gene in their DNA, one from each parent. Copies of the same gene with different mutations are called alleles . When parents make a sperm or an egg, alleles in each parent are shuffled and recombined, and only one allele of a gene ends up in each sperm or egg cell. When the reshuffled alleles from a mother and a father are combined when sperm and eggs join, new mixes of alleles are created in the babies [ 2 , 3 ]. The mixing of alleles allows for new combinations of mutations and characteristics, adding to a species’ genetic diversity ( Figure 2 ).

- Figure 2 - (A) Genetic diversity is generated when mutations create new alleles over time.
- Mixing alleles from parents creates new combinations of alleles in their babies. Organisms that can clone themselves, like bacteria, can pass alleles to each other. Each coloured dot represents a different allele. (B) Genetic diversity can be lost when habitat loss divides populations or when buildings or highways isolate populations. (C) Creating protected areas where individuals from different populations can migrate and spread their genes can help a species to maintain its genetic diversity.
Not all species need a mother and a father to make a baby. Bacteria can clone themselves ( Figure 2 ) and directly pass their alleles from a parent to its identical clone [ 3 ]. Any mistakes in the parent’s DNA will be passed on to the clone. Amazingly, bacteria can also give alleles to each other, even if they are not related! This is a unique way simple species like bacteria can increase their genetic diversity, without relying on the mixing of alleles between a mother and a father [ 4 ].
Why is Genetic Diversity Important?
When a species has a lot of differences in its DNA, we say that genetic diversity is high [ 2 ]. In species with high genetic diversity, there are lots of mutations in the DNA, which cause differences in the way individuals look as well as differences in important traits that we cannot see [ 2 ]. This is called adaptation . For example, some types of apples can grow better in hotter environments, thanks to their genes. The variety of characteristics in species with high genetic diversity means they are more likely to successfully cope with changes in their environment. A great example of this is seen in the peppered moths during the industrial revolution [ 4 ]. Natural genetic diversity in peppered moths produced different wing colours, ranging from light to dark. Before the Industrial Revolution, peppered moths with light wings were more common because they had the best camouflage on white tree trunks. The Industrial Revolution caused a lot of air pollution that started to cover tree trunks, making them black. Light-winged moths were no longer camouflaged and were easy prey for birds. But dark-winged individuals were now hidden! This meant that dark moths had an advantage and were more likely to live long enough to have babies. The babies of dark moths were also dark because of the alleles they inherited from their parents, so they were also more likely to survive. The dark moths had higher fitness and became more common as a result [ 4 ].
What Happens When Genetic Diversity is Low?
When few mutations are found in the DNA of a species, genetic diversity is said to be low [ 2 ]. Low genetic diversity means that there is a limited variety of alleles for genes within that species and so there are not many differences between individuals. This can mean that there are fewer opportunities to adapt to environmental changes. Low genetic diversity often occurs due to habitat loss. For example, when a species’ habitat is destroyed or broken up into small pieces, populations become small. Small, fragmented populations can lead to loss of genetic diversity because fewer individuals can survive in the remaining habitat so fewer individuals breed to pass on their alleles. In small populations, the choice of mates is also limited. Over time, individuals will all become related and will be forced to mate with relatives. This is inbreeding . Inbred animals often have two identical alleles for their genes because the same gene was passed on from both parents. If this allele has harmful mutations, an inbred baby can be unhealthy. This is called inbreeding depression [ 2 ].
If genetic diversity gets too low, species can go extinct and be lost forever. This is due to the combined effects of inbreeding depression and failure to adapt to change. In such cases, the introduction of new alleles can save a population. This is called genetic rescue [ 2 ]. In the 1990s conservation scientists had to use genetic rescue to save the Florida panther, which was threatened by extinction due to low genetic diversity ( Figure 3 ) [ 5 ]. Very few Florida panthers remained and their genetic diversity was extremely low. Many Florida panther babies were sick because of inbreeding depression. A closely related panther with high genetic diversity was present in Texas. Texan panthers were moved to Florida to have babies with the Florida panthers. This increased genetic diversity because of the mixing of alleles we spoke about before. Soon after the Texan panthers arrived, many healthy kittens were born [ 5 ].

- Figure 3 - (A) The Florida panther was once widespread, with high genetic diversity.
- (B) Hunting and habitat loss reduced population size and resulted in very low genetic diversity and inbreeding. (C) Eight female panthers from Texas were moved to Florida to breed with Florida panthers. (D) When the Texas and Florida panthers bred, new alleles were introduced into the population, helping the Florida panther population become bigger and healthier over time.
What’s Happening to Genetic Diversity Around the World?
We hear a lot about the loss of species in the world, but we are also seeing a loss of genetic diversity within species. The increasing number of people on Earth and our increasing use of natural resources has reduced space and resources for wild species. Over time, many wild animal and plant populations have become smaller or more isolated. Many species have also gone through local extinctions. This has led to a global loss of genetic diversity. Scientists think that the genetic diversity within species may have declined by as much as 6% globally since the Industrial Revolution [ 6 ]. This means that many species are less able to adapt when facing new challenges, like climate change, pollution, and new diseases. If too much genetic diversity is lost, more and more species could become unhealthy and in need of conservation actions similar to the Florida panther. However, there are steps we can take to conserve and restore genetic diversity across many species.
How Do We Stop Genetic Diversity Loss?
We must preserve and protect genetic diversity. This can be done through the conservation of our remaining wild populations [ 2 ]. We can use nature reserves and wildlife bridges to reconnect wild populations that have become separated by our cities and highways. We can also restore habitats, because this will allow wild populations to get bigger. Sometimes we can even remove harmful stressors and pests so that populations can naturally regrow. We can also reintroduce species that have been lost from habitats they used to live in. Taken together, these strategies can help stop genetic diversity loss. It is important to protect genetic diversity because it is the foundation for healthy species. Healthy species are necessary for human health and for the health of the whole planet!
Gene : ↑ A section of DNA that contains the instructions for a trait.
Genetic Diversity : ↑ The overall diversity in the DNA between the individuals of a species.
Mutation : ↑ A change in an organism’s DNA. This can be a change of a single letter or a much bigger change of hundreds of letters at once.
Adaptation : ↑ The process of a species changing in order to better survive in its environment.
Alleles : ↑ Different variations of a gene caused by mutations. Many species have two alleles for every gene, one copy from each parent.
Inbreeding : ↑ Breeding between closely related individuals. Inbreeding often happens when populations are small and there are few options for mating. Inbred individuals are usually less healthy.
Inbreeding Depression : ↑ Inbred individuals share ancestors and are more likely to have identical copies of genes. If these genes contain harmful mutations, they will be expressed and cause lower health of inbred individuals.
Genetic Rescue : ↑ A conservation strategy, new individuals are moved into a population to increase genetic diversity and improve population health.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
[1] ↑ Meyer, R., and Purugganan, M. 2013. Evolution of crop species: genetics of domestication and diversification. Nat. Rev. Genet. 14:840–52. doi: 10.1038/nrg3605
[2] ↑ Frankham, R., Ballou, J. D., and Briscoe, D. A. 2002. Introduction to Conservation Genetics. Cambridge: Cambridge University Press. p. 617.
[3] ↑ Emamalipour M., Seidi K., Zununi V. S., Jahanban-Esfahlan A., Jaymand M., Majdi H., et al. 2020. Horizontal gene transfer: from evolutionary flexibility to disease progression. Front. Cell. Dev. Biol. 8:229. doi: 10.3389/fcell.2020.00229
[4] ↑ Cook, L. M., and Saccheri, I. J. 2013. The peppered moth and industrial melanism: evolution of a natural selection case study. Heredity 110:207–12. doi: 10.1038/hdy.2012.92
[5] ↑ Johnson, W. E., Onorato, D. P., Roelke, M. E., Land, E. D., Cunningham, M., Belden, R. C., et al. 2010. Genetic restoration of the Florida panther. Science . 329:1641–5. doi: 10.1126/science.1192891
[6] ↑ Leigh, D. M., Hendry, A. P., Vázquez-Domínguez, E., and Friesen, V. L. 2019. Estimated six per cent loss of genetic variation in wild populations since the industrial revolution. Evol. Appl. 12:1505–12. doi: 10.1111/eva.12810
MIT Libraries home DSpace@MIT
- DSpace@MIT Home
- MIT Libraries
- Doctoral Theses
Essays on genetic variation and economic behavior

Other Contributors
Terms of use, description, date issued, collections.
- Skip to primary navigation
- Skip to main content
- Skip to primary sidebar
- Skip to footer
- Image & Use Policy
- Translations
UC MUSEUM OF PALEONTOLOGY
Understanding Evolution
Your one-stop source for information on evolution
- ES en Español
Mutations are changes in the information contained in genetic material. For most of life, this means a change in the sequence of DNA, the hereditary material of life. An organism’s DNA affects how it looks, how it behaves, its physiology — all aspects of its life. So a change in an organism’s DNA can cause changes in all aspects of its life.
Mutations are random Mutations can be beneficial, neutral, or harmful for the organism, but mutations do not “try” to supply what the organism “needs.” In this respect, mutations are random — whether a particular mutation happens or not is unrelated to how useful that mutation would be.
Not all mutations matter to evolution Since all cells in our body contain DNA, there are lots of places for mutations to occur; however, not all mutations matter for evolution. Somatic mutations occur in non-reproductive cells and so won’t be passed on to offspring.
For example, the yellow color on half of a petal on this red tulip was caused by a somatic mutation. The seeds of the tulip do not carry the mutation. Cancer is also caused by somatic mutations that cause a particular cell lineage (e.g., in the breast or brain) to multiply out of control. Such mutations affect the individual carrying them but are not passed directly on to offspring.

The only mutations that matter for the evolution of life’s diversity are those that can be passed on to offspring. These occur in reproductive cells like eggs and sperm and are called germline mutations .
- More Details
- Evo Examples
- Teaching Resources
Read more about how mutations are random and the famous Lederberg experiment that demonstrated this. Or read more about how mutations factored into the history of evolutionary thought . Or dig into DNA and mutations in this primer.
Learn more about mutation in context:
- Evolution at the scene of the crime , a news brief with discussion questions.
- A chink in HIV's evolutionary armor , a news brief with discussion questions.
Find lessons, activities, videos, and articles that focus on mutation.
Reviewed and updated June, 2020.
Genetic variation
The effects of mutations
Subscribe to our newsletter
- Teaching resource database
- Correcting misconceptions
- Conceptual framework and NGSS alignment
- Image and use policy
- Evo in the News
- The Tree Room
- Browse learning resources
- Share full article
Advertisement
Supported by
The Ever-Resilient Pupfish Makes a Comeback in Death Valley
The spring population of the critically endangered species is at a 25-year high, a surprising rebound in a tiny desert cave.

By Alexander Nazaryan
When it comes to sheer resilience, few, if any, species can match the tiny Devils Hole pupfish.
Cyprinodon diabolis , as the species is known, has the most ruthlessly circumscribed natural habitat of any vertebrate: Devils Hole , an exceptionally deep, water-filled cave in a limestone formation in the unforgiving Nevada desert, where the fish mostly stay on a rock shelf little more than 200 square feet. Not only that, but the pupfish are believed to be one of the most inbred of all species , a lack of genetic variation that makes it difficult for the creatures to procreate and thrive.
And yet, improbably, Devils Hole pupfish are thriving. Late last month, the National Park Service announced that the spring population of the species had grown to 191 , the highest in 25 years, according to a count conducted twice a year by scuba divers. Because of seasonal fluctuations in food sources, fall counts tend to be higher, meaning that this year’s tally could be a watershed.
“If, this fall, we have over 300, I’ll be really ecstatic,” said Kevin Wilson, an aquatic ecologist at the National Park Service who has studied the Devils Hole pupfish for more than two decades. (Devils Hole is officially part of Death Valley National Park , most of which is in California.)
If the pupfish census does not seem especially impressive, consider that there were only 35 pupfish left in Devils Hole in 2013, prompting worries about extinction. For now, that danger has receded ever so slightly.
“This is a tremendous success story,” said Christopher Martin , an evolutionary biologist and pupfish expert at the University of California, Berkeley. “Ten years ago, we couldn’t have expected this level of success.”
Biologists have been feeding the pupfish frozen food to supplement their regular diet of algae since 2007. In 2019, the biologists finally arrived at the optimal formula of mysid shrimp, water fleas and blood worms. “This change in the supplemental food probably did enhance that increase in population numbers we’re seeing,” Dr. Wilson said.
Hurricane Hilary, which hit last summer, also helped. Even though the storm caused flooding and damage to the park , it benefited the pupfish living in Devils Hole by “adding nutrients that washed off the surrounding land surface in a fine layer of clay and silt,” according to the National Park Service.
The tiny pupfish, usually about an inch in size, is believed to have lived in Devils Hole for at least 10,000 years and probably much longer, Dr. Martin said. Its name alludes to a playful, puppylike disposition.
How the pupfish ended up in the Nevada desert is not known for certain. Much of Nevada was once underwater . The waters eventually receded, but somehow the pupfish found a refuge in the vast expanse of scrubland and sand.
To this day, no person is known to have completed an exploration of the lowest depths of Devils Hole, which is hundreds of feet deep. (A submersible would never fit into the narrow cavern, Dr. Wilson said.) In a notorious accident in 1965, two young men died during a dive in Devils Hole .
Not much for deepwater exploration, the pupfish stay at depths of 80 feet or less. There, the temperature is 93 degrees Fahrenheit, potentially even hotter near the surface.
Because the pupfish are effectively perched on a shallow underwater ledge, changes to the water table can harm prospects for survival. Dr. Wilson worries that the profusion of enormous solar panel farms in the surrounding desert could drastically increase water usage, damaging the delicate Amargosa River system . Mining is booming again in Nevada. Pahrump, a desert town near Ash Meadows, has seen its population explode .
“There’s increasing pressure on groundwater,” Dr. Wilson said. A well could inadvertently tap the Devils Hole aquifer, causing a drastic drop in the water level there.
Perhaps the greatest danger to the species is its lack of genetic diversity, which increases the incidence of harmful genetic mutations and thus makes it harder for the population to grow. In a classic Catch-22, the pupfish have only one way of inbreeding less: by growing their population.
To prepare for potential catastrophe, the U.S. Fish and Wildlife Service has been breeding Devils Hole pupfish in captivity since 2013. Introducing bred pupfish into Devils Hole is unfeasible for a variety of reasons, but should something happen to the wild pupfish, the species will live on.
For now, however, the wild pupfish are hanging on in Devils Hole. Christopher Norment, a vertebrate ecologist and the author of “Relicts of a Beautiful Sea,” a book about Death Valley, said that although he was “somewhat jaundiced” about the long-term prospects of the Devils Hole pupfish, he was impressed by its tenacity.
“It’s the story of survival in the face of overwhelming odds,” he said.
Explore the Animal Kingdom
A selection of quirky, intriguing and surprising discoveries about animal life..
Scientists say they have found an “alphabet” in the songs of sperm whales , raising the possibility that the animals are communicating in a complex language.
Indigenous rangers in Australia’s Western Desert got a rare close-up with the northern marsupial mole , which is tiny, light-colored and blind, and almost never comes to the surface.
For the first time, scientists observed a primate in the wild treating a wound with a plant that has medicinal properties.
A new study resets the timing for the emergence of bioluminescence back to millions of years earlier than previously thought.
Scientists are making computer models to better understand how cicadas emerge collectively after more than a decade underground .
COMMENTS
Genetic variation refers to differences among the genomes of members of the same species. A genome is all the hereditary information—all the genes—of an organism.For instance, the human genome contains somewhere between twenty and twenty-five thousand . genes.. Genes are units of hereditary information, and they carry instructions for building proteins.
Variable segments of DNA that are 3, 4, or 5 bases long, repeated over and over. Single nucleotide polymorphism (SNP, pronounced, "snip"). A variation in one base pair at a particular location in the genome. SNPs are useful in comparing genetic variations between individuals and in populations.
Evo 101. Genetic variation. Without genetic variation, some key mechanisms of evolutionary change like natural selection and genetic drift cannot operate. There are three primary sources of new genetic variation: Mutations are changes in the information contained in genetic material. (For most of life, this means a change in the sequence of DNA.)
Genetic variation refers to differences in the genetic makeup of individuals in a population. Genetic variation is necessary in natural selection. In natural selection, organisms with environmentally selected traits are better able to adapt to the environment and pass on their genes. Major causes of variation include mutations, gene flow, and ...
Figure 19.2A. 1 19.2 A. 1: Low genetic diversity in the wild cheetah population: Populations of wild cheetahs have very low genetic variation. Because wild cheetahs are threatened, their species has a very low genetic diversity. This low genetic diversity means they are often susceptible to disease and often pass on lethal recessive mutations ...
Genetics is the scientific study of inherited variation.Human genetics, then, is the scientific study of inherited human variation.. Why study human genetics? One reason is simply an interest in better understanding ourselves. As a branch of genetics, human genetics concerns itself with what most of us consider to be the most interesting species on earth: Homo sapiens.
Genetic diversity influences biodiversity, and thus NCP, in two main ways: (1) through standing genetic variation (that is, the particular combination of genes and alleles present at a given time ...
The establishment of a new technology (genome sequencing) enabled the measurement of a signal that is informative about disease risk (genetic variation) but is also influenced by evolutionary history.
From subtle shifts in the genetic makeup of a single population to the entire tree of life, evolution is the process by which life changes from one generation to the next and from one geological epoch to another. The study of evolution encompasses both the historical pattern of evolu-tion—who gave rise to whom, and when, in the tree of life ...
Human genetic variants are typically referred to as either common or rare, to denote the frequency of the minor allele in the human population. Common variants are synonymous with polymorphisms ...
A 3-panel cartoon, each showing a hawk flying above a group of mice. In the first panel there are 3 black mice and 6 tan mice. The black mice match the black ground. Text reads A population of mice has moved into a new area where the rocks are very dark. Due to natural genetic variation, some mice are black, while others are tan.
Genetic variation is the difference in DNA among individuals or the differences between populations among the same species. The multiple sources of genetic variation include mutation and genetic recombination. Mutations are the ultimate sources of genetic variation, but other mechanisms, such as genetic drift, contribute to it, as well.. Darwin's finches or Galapagos finches Parents have ...
Evolution - Genetic Variation, Rate, Adaptation: The more genetic variation that exists in a population, the greater the opportunity for evolution to occur. As the number of gene loci that are variable increases and as the number of alleles at each locus becomes greater, the likelihood grows that some alleles will change in frequency at the expense of their alternates. The British geneticist R ...
In fact, contemporary whole genome genetic variation data emphasize the greater imprint of demography than biology in our genomes. ... As the essays by Richard Cooper and Troy Duster eloquently argue, the nongenetic factors in human disease, equally if not more importantly, affect our genetic biology in fundamental ways, and in many cases merit ...
6.2 Summary. No two human individuals are genetically identical (except for. monozygotic. twins), but the human species as a whole exhibits relatively little genetic diversity, relative to other mammalian species. Genetically, two people chosen at random are likely to be 99.9 per cent identical.
Fertilization is the fusion of gametes from two parents. Fertilization leads to new, diploid offspring. Sexual reproduction results in genetic variation, or genetic differences, between parents and offspring. Offspring inherit one set of chromosomes from each parent. So, an offspring has a mixture of chromosomes (and alleles) from its two parents.
Gene: ↑ A section of DNA that contains the instructions for a trait. Genetic Diversity: ↑ The overall diversity in the DNA between the individuals of a species. Mutation: ↑ A change in an organism's DNA. This can be a change of a single letter or a much bigger change of hundreds of letters at once.
ADVERTISEMENTS: Here is an essay on 'Genetic Variation' for class 9, 10, 11 and 12. Find paragraphs, long and short essays on 'Genetic Variation' especially written for school and college students. Essay on Genetic Variation Essay # 1. Meaning of Genetic Variation: Evolution requires genetic variation. If there were no dark moths, the population could […]
Abstract. This thesis is a collection of papers in which behavior genetic methods are used to shed light on individual differences in economic preferences, behaviors and outcomes. Chapter one uses the classical twin design to provide estimates of genetic and environmental influences on experimentally elicited preferences for risk and giving.
Genetic variation is transferred from one generation to the next and it generates phenotypic variation within a species population. The primary source of genetic variation is mutation (changes in the DNA base sequence) Mutation results in the generation of new alleles. The new allele may be advantageous, disadvantageous or have no apparent ...
genetic variation. In many edited books covering diverse topics, major emphasis is placed toward presenting landscape essay on the entire scope of the subject. It is difficult to present all aspects of genetic variation, which is discussed in quantitative, popula-tion, and evolutionary genetics, in one volume, but a good
AQA A-Level Biology - Cause and Importance of Variation and Diversity Essay. Difference in DNA leads to genetic diversity. Click the card to flip 👆. - Crossing over of alleles = homologous pairs associate, chiasmata form, alleles are exchanged, new combination of alleles. - Independent segregation = alleles of two (or more) different genes ...
Evo 101. Mutations. Mutations are changes in the information contained in genetic material. For most of life, this means a change in the sequence of DNA, the hereditary material of life. An organism's DNA affects how it looks, how it behaves, its physiology — all aspects of its life. So a change in an organism's DNA can cause changes in ...
The spring population of the critically endangered species is at a 25-year high, a surprising rebound in a tiny desert cave. By Alexander Nazaryan When it comes to sheer resilience, few, if any ...